Introduction
In the past decade, the synthesis of functional
nano-architectures, aimed at developing electronic and photonic nano-devices to
perform specific functions, such as catalysis, chemical sensing, electrical
conductivity, photodynamic
therapy, etc., has been an intense area of research [1-10].
Most of these activities are driven by the self-assembly and self-organization
properties of molecules and require the incorporation of functional building
blocks through a well-defined controlled process. Porphyrins
(Por), tetraazaporphyrins, Phthalocyanines
(Pc) and porphyrazines are examples of tetrapyrrole macrocyclic ring systems,
through which mutual non-covalent recognition of specific structural
properties, can self-assemble and act as building blocks for self-organized
architectures.
Phthalocyanines
possess an extended flat hydrophobic aromatic surface and the disc-shaped rigid
Pc rings can easily stack through strong intermolecular arene-arene (π-π)
interactions, leading to aggregation [11-13]. Other molecular recognition
motifs that can self-aggregate are metal-ligand and donor-acceptor interactions
and hydrogen bonding [14-16]. Various approaches have been explored in order to
control the self-organization of Pc molecules to form well-defined
nano-objects. One such approach is the incorporation of these molecules in
amphiphilic polymers [17-20].
Pcs have been incorporated in such systems as a side
group, as a terminal group, in the main chain and in a polymeric network [21-26].
Kimura et al studied the self-assembly properties of ZnPc-terminated Butyl
Acrylate (BA) and Tri(Ethylene Glycol)Methyl Ether Acrylate (TEGA) polymers and
have reported formation of nanofibers through self-organization [18]. This
group also studied the aggregation behavior of the amphiphilic block copolymer of
poly(norbornene)s containing Pc moieties as a side chain of the polymer
backbone that formed spherical micelles in an alkaline aqueous solution [19]. In
addition, ordered stacked Pc polymers were prepared in which the Pc moieties
were contained in the polymer network and in the main chain of the Pc polymers
with reported rod-like nanostructures [20]. Similar polymer systems have also
been developed using Por moieties [27-30]. In most cases, the polymerization
step has been achieved by controlled/living radical polymerization, such as
metathesis polymerization and Atom Transfer
Radical Polymerization (ATRP) [18-20, 27-30].
Pcs have mostly been incorporated in amphiphilic
polymers as a side group, a terminal group, or in the main chain [31-34]. Although
polymer amphiphiles with Por cores have been designed and reported, amphiphilic
polymers especially block copolymers with Pc cores; have not been studied well
due to synthetic challenges. We report here the synthesis and characterization
of a new di-block acrylate copolymer with CuPc core, having a distinct
hydrophobic segment composed of tert-Butyl
Acrylate (tBA) units, and a hydrophilic segment having Tri(Ethylene
Glycol)Monomethyl Ether Acrylate (TEGA) units.
Experimental
Materials
and Methods
Unless mentioned otherwise, all reagents were used as
received from commercial suppliers without further purification. Dimethylformamide
(DMF) used in these reactions was purchased as anhydrous grade stored in a
Sure-Seal™ bottle. Toluene was dried by distillation over freshly
cut sodium metal cubes. All reactions involving moisture-and air-sensitive
reagents were carried out under an inert atmosphere using high-purity grade
argon that was first passed through a column of anhydrous calcium sulfate. Thin Layer
Chromatography (TLC) was done on 60, 200 μm thick
silica gel flexible plates and 250 μm aluminum oxide flexible plates. Gravity-flow
column chromatography was performed on 70-230-mesh, 60Å
silica gel or 150-mesh, 58-Å activated neutral aluminum oxide.
Structural characterization by Fourier-Transform
Infrared spectroscopy (FT-IR) was carried out using a Thermo
Nicolet Nexus 470 FT-IR spectrometer and a Perkin-Elmer System 2000 instrument.
The electronic absorption spectra were recorded on a Perkin Elmer (UV/VIS/NIR)
spectrometer Lambda-19. 1H-NMR and 13C-NMR spectra were
recorded by a 300 MHz Bruker instrument. Elemental analyses were performed on a
Perkin-Elmer Series II CHNS/O Analyzer 2400 machine. Scanning Electron
Microscopy (SEM) was performed by a JEOL JSM5900-LV instrument
and Atomic Force Microscopy (AFM) by Picoscan 3000 AFM (Agilent Technology).
Synthesis
of the Phthalocyanine Precursor, 2,3-Dicyano-1,4-bis {2-2-[(2-
hydroxyethoxy)-ethoxy]-ethoxy}benzene (1)
2,3-dicyano-1,4-hydroquinone (11.3958 g, 71.1659
mmol) in 100 mL anhydrous DMF was added to a stirred suspension of K2CO3
(29.5075 g, 213.5 mmol) and KI (2.9834 g, 18 mmol) in 150 mL anhydrous DMF
under inert atmosphere in a 500 mL 3 neck round bottom flask. A solution of
2-[2-(2-chloroethoxy)-ethoxy]ethanol (30 g, 170.7982 mmol) in 10 mL anhydrous
DMF was added to it. The reaction mixture was stirred at 70oC for 10
days. It was then cooled to room temperature and filtered under vacuum. The
filtrate was poured into a Teflon petridish and DMF was allowed to evaporate. When
it was dry, it was kept in the oven at 50oC under vacuum for 2
hours. The brown solid was stirred with water and extracted with chloroform. The
organic layer was collected, dried over anhydrous MgSO4, filtered
and chloroform was evaporated in a rotary evaporator to give orange colored
solid. The solid was purified by column chromatography, using alumina column,
eluting with first with Dichloromethane (DCM),
followed by 5% methanol in DCM. Evaporation of the solvent yielded compound 1
as white solid Yield: 70%.
FT-IR (KBr): ν=3410, 2908, 2876, 2227, 1493, 1352,
1285, 1195, 1102, 1065, 925, 820, 738, 523, 469 cm-1; 1H
NMR (300 MHz, DMSO-d6 δ=2.5): δ=7.62 (m, 2H), δ=4.56 (t, 1H), δ=4.28
(t, 4H), δ= 3.77 (t, 4H), δ=3.59-3.62 (m, 4H), δ=3.51-3.54 (m, 4H), δ=3.46 (t,
4H), δ=3.39-3.42 (m, 4H).
Synthesis
of Copper Phthalocyanine (CuPc) (2)
1 (2.682 g, 6.32 mmol) and copper powder (0.8 g,
12.59 mmol) were taken in a 10 mL round bottom flask and heated for 5 hours at
170oC under argon. The color of the reaction mixture turned green. The
reaction mixture was cooled to room temperature and 10 mL of methanol was added
to it. It was stirred overnight and filtered under vacuum. Methanol was
evaporated in a rotary evaporator to give green solid. It was purified by
column chromatography using alumina. The solvent systems used were DCM,
followed by 2-5% methanol in DCM. TLC showed that the product is still not
pure. So the fractions that eluted with 2-5% methanol in DCM, were combined and
subjected to another column chromatography, with a solvent system of DCM, again
followed by 2-5% methanol in DCM. The fractions obtained with the last eluent
system were checked with TLC and were found to be pure. Solvent was evaporated
to give pure CuPc (2) as a sticky green solid. Yield: 2.2 g, 20%. Anal. Calcd.
for C83H123CuN8O32 (%): C, 55.1; H,
6.9; N, 6.2. Found (%): C, 54.9; H, 6.8; N, 6.3. FT-IR (KBr): ν = 3391, 2901,
2874, 1601, 1505, 1460, 1317, 1268, 1213, 1096, 1068, 924, 890, 808 cm-1;
1H NMR (300 MHz, DMSO-d6 δ= 2.5): δ = 7.4 (br, 8H), δ = 4.25 (t,
8H), δ = 3.91 (t, 16H), δ = 3.6-3.8 (m, 80H); UV-Vis (DMSO): λmax =
741, 662, 325, 269 nm.
Synthesis
of ATRP macroinitiator (3)
Compound 2 (0.5 g, 0.2765 mmol) was dissolved in 8
mL pyridine in a 25 mL round bottom flask in presence of argon. 2-Bromoisobutyryl
bromide (7 g, large excess) was dissolved in 5 mL chloroform and added drop
wise to a vigorously stirred solution of 11 at 0oC over a period of
1 hour under inert atmosphere. The temperature gradually rose to room
temperature and the reaction was allowed to proceed for 24 hours. Chloroform
was evaporated in a rotary evaporator. The reaction mixture was then added drop
wise to a large excess of water when green solid separated out. It was washed
thoroughly with hexane in order to remove unreacted 2-bromoisobutyryl bromide. It
was then extracted with chloroform and the organic layer was washed thoroughly
with water, a saturated solution of K2CO3 and a saturated
solution of NaCl, twice each.
The organic layer was collected, dried over
anhydrous MgSO4, and chloroform was evaporated. The green solid was
purified by column chromatography using alumina column and chloroform as the
eluent. Evaporation of chloroform in a rotary evaporator yielded 12 as a dark
green sticky solid. Yield: 0.3 g, 40%. Anal. Calcd. for C115H163Br8CuN8O40
(%): C, 46; H, 5.5; N, 3.7. Found (%): C, 45.01; H, 5.4; N, 3.6. FT-IR (KBr): ν
= 2872, 1732, 1599, 1502, 1462, 1388, 1371, 1272, 1211, 1167, 1107, 1073, 928,
875, 756 cm-1; 1H-NMR (300 MHz, DMSO-d6 δ=
2.5): δ = 7.4-7.6 (m, 8H), δ = 4.22 (t, 16H), δ = 3.3-3.76 (m, 80H), δ = 1.86
(s, 48H); λmax = 742, 664, 326, 244 nm.
Synthesis
of the homopolymer, CuPc-core-poly-(tBA) (4a)
The macroinitiator 3 (0.11 g, 0.0367 mmol) was
dissolved in dry toluene for 30 minutes under argon atmosphere. N, N, N', N',
N"-Pentamethyldiethylenetriamine
(PMDETA) (0.063 mL, 0.301 mmol), the monomer t-BA (5 times the weight of the
monomer) and Cu(I)Br (0.0432 g, 0.301 mmol) were added sequentially. Three
freeze-pump-thaw cycles were performed. The reaction mixture was then heated
under vacuum at 90oC for 4 hours under argon atmosphere. Toluene was
evaporated. The mixture was washed several times with hexane to remove
unreacted PMDETA and the monomer. After each washing, it was centrifuged and
hexane was decanted. The green solid was dissolved in chloroform and filtered. The
residue is unreacted Cu(I)Br. The filtrate was collected and chloroform
evaporated to give green solid.
FT-IR (KBr): ν=2921, 2873, 1732, 1496, 1463, 1388,
1371, 1276, 1168, 1108, 1069, 957, 874, 814, 762, 701, 611 cm-1; 1H-NMR
(300 MHz, CDCl3 δ=7.26): δ=4.22 (t, 16H), δ=3.6-3.7 (m, 80H), δ=2.2
(br, CH of the polymer backbone), δ=1.9 (br, CH2 of the polymer
backbone), δ=1.43 (br, (CH3)3C of the t-BA ester group); UV-Vis
(DCM): λmax=741, 663, 325, 231 nm.
Synthesis
of the block copolymer, CuPc-core- poly-(tBA)-co-poly-(TEGA) (4b)
4b was made following the same procedure as the
synthesis of 4a. In this case, 4a itself was used as the macroinitiator for the
polymerization reaction.
1H-NMR (300 MHz,
CDCl3 δ=7.26): δ=7.6 (m, 8H), δ=4.3 (t, 16H, CH2OCO), δ=3.5-3.75
(m, OCH2), δ=3.38 (s, CH3 of the TEGacrylate group), δ=2.2-2.5
(br, m, protons in polymer backbone), δ=1.44 (s, (CH3)3C of the t-BA
ester group); UV-Vis (DCM): λmax=737, 292 nm.
Synthesis
of TEGA monomer (5)
Tri(ethylene glycol) monomethyl ether (1.5 g, 9.1352
mmol) was dissolved in 5 mL anhydrous THF. Acryloyl chloride (1.5 g, 16.5727
mmol) and triethyl amine (3.18 mL, 22.838 mmol) were added to it. The reaction
mixture was stirred under argon atmosphere overnight at room temperature. It
was then poured into water and extracted with chloroform. The organic layer was
washed several times with a saturated solution of Na2CO3
and a saturated solution of NaCl.
It was dried over anhydrous MgSO4,
filtered and chloroform was evaporated from the filtrate to yield the TEGA
monomer (5) as colorless liquid. 5 were purified by column chromatography using
alumina. 2% methanol in chloroform was used to elute the pure fraction.
FT-IR (KBr): 2872, 1722, 1631, 1455, 1411, 1269,
1195, 1113, 987, 853, 806 cm-1; 1H-NMR (300 MHz, CDCl3
δ=7.26): δ=6.35 (dd, 1H), δ=6.1 (dd, 1H), δ=5.79 (dd, 1H), δ=4.25 (t, 2H), δ=3.46-3.7
(m, 10H), δ = 3.33 (s, 3H).
Results
and Discussion
Retro-synthetic
Analysis
The two key components of the retro-synthetic design
for this project are (i) an appropriate core molecule functionalized with
initiator groups and (ii) polymerization to form the arms. Therefore, in order
to make CuPc core functionalized polymer arms (4a and 4b), a CuPc derivative
containing eight bromoisobutyryl initiator moieties (3) must be synthesized
first. This macro-initiator could be made from a CuPc-centered
tri(ethyleneoxy) intermediate (2), a CuPc derivative
with peripherally substituted hydroxyl (-OH) functional groups. That leaves 2
was to be constructed by cyclo-tetramerization of a phthalonitrile precursor (1).
Synthesis
and Characterization
Phthalonitriles are the most commonly used
precursors for the synthesis of phthalocyanine derivatives. A disubstituted
phthalonitrile derivative (1) was synthesized as the precursor of 2 by an
O-alkylation reaction in which 2,3-dicyanohydroquinone was alkylated by
treatment with 2-[2-(2-chloroethoxy)-ethoxy]ethanol in the presence of
anhydrous potassium carbonate and a catalytic amount of potassium iodide (Scheme 1). The reaction was carried out
in anhydrous DMF. The same reaction conditions were followed as reported by Xue
et al., [16]. After evaporation of DMF, the crude product was extracted with
chloroform and purified by column chromatography to afford pure 1 as a white
solid.
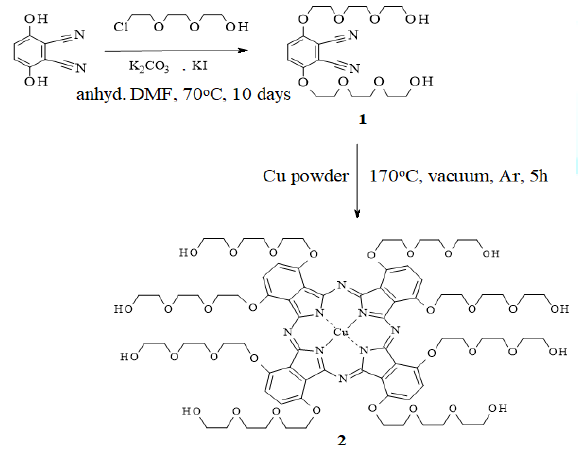
Scheme1:Synthesis of CuPc precursor (1) andCuPc (2).
The first attempt to synthesize CuPc was by
refluxing 1 in 2-(dimethylamino)ethanol in the presence of copper acetate under
an argon atmosphere, but no characteristic green color of Pc was obtained, even
after 3 days. Next, we tried to synthesize the CuPc via its Li2Pc
[35]. After purification, the desired CuPc was obtained, but in very low yield
(12%). The next attempt was via the isoindoline route where 1 was first
converted into its isoindolene by treating with ammonia in the presence of
sodium methoxide in methanol, followed by self-condensation of the isoindolene
into CuPc, in the presence of copper acetate, but without success. Another
attempted route to 2 was by reacting 1 with copper acetate in dry quinoline at
220oC for 3 hours, but the characteristic green color of Pc was not
observed. Finally, we succeeded in synthesizing 2 from 1 by a solvent-free
method [36]. The phthalonitrile derivative was stirred with Cu powder at a very
high temperature [36]. The optimum temperature for our system was found to be
170oC under vacuum for 5 hours (Scheme 1). Unreacted Cu powder was
filtered off and pure 2 were obtained as a sticky green solid after column
chromatography. The macroinitiator 3, with bromoisobutyryl
initiating sites, was synthesized by acylation of the peripheral hydroxyl
groups in 1 with a large excess of 2-bromoisobutyryl bromide. The reaction was
carried out in a mixture of chloroform and pyridine and the temperature was
allowed to rise from 0oC to room temperature and maintained there
for 24 hours (Scheme 2). The crude
product was extracted with chloroform and washed thoroughly with saturated
solutions of Potassium Carbonate (K2CO3) and Sodium
Chloride (NaCl). Pure 3 was obtained as a bright green solid after column
chromatography.

Scheme2:Synthesis of CuPc macroinitiator (3).
The polymerization reactions were performed under
ATRP conditions (Scheme-3). Two
monomers were chosen: (i) tert-Butylacrylate (t-BA) and (ii) Tri(ethylene
glycol)Monomethyl Ether Acrylate (TEGA). While the polymer chain composed of
t-BA units imparts hydrophobicity to the system, the one composed of TEGA
monomers provides hydrophilicity. Accordingly, the CuPc-core block copolymer
poly-(t-BA)-poly-(TEGA) will be amphiphilic in nature. The monomer TEGA (5) was
obtained from tri(ethylene glycol)monomethyl ether and acryloyl chloride
following the conditions previously reported for the preparation of acrylated
PEG derivatives [37]. Copper (I) Bromide [Cu(I)Br] was used as catalyst and
N,N,N',N',N"-Pentamethyldiethylenetriamine (PMDETA) as the ligand. In a
typical polymerization procedure, the Pc-core macroinitiator: CuBr: ligand
ratio was 1: 8.2: 8.2 and the amount of monomer added was 5 times the weight of
the initiator. Polymerization reactions were carried out in solution with
toluene as solvent under argon atmosphere. The purification procedure was
modified, by first extracting with chloroform and then purified by column
chromatography.
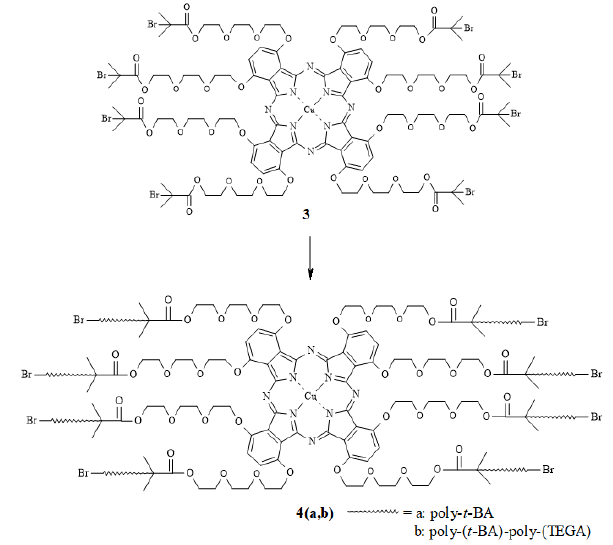
Scheme3:Polymerization of t-BA and TEGA.by ATRP.
The FT-IR spectrum of the phthalonitrile precursor (1) showed strong O-H stretching at 3410
cm-1 and aromatic C-H stretch at 3092 cm-1. Bands at 2907
cm-1 and 2876 cm-1 are indicative of CH2
asymmetric and CH2 symmetric stretching frequencies. The strong peak
at 2228 cm-1 is typical of C≡N stretching in aromatic nitriles. The
presence of both aromatic and aliphatic C-H vibrations proves that the
O-alkylation step was been successful. The C≡N peak at 2228 cm-1 was
completely disappeared in the spectrum of CuPc (2), indicating complete cyclotetramerization of 1. The strong peak
at 3390 cm-1 is due to O-H stretching. The aromatic C-H stretching
vibration is seen at 3080 cm-1 and the positions of symmetric and
asymmetric aliphatic C-H stretching vibrations remain the same as those for
compound 1. In the FTIR spectrum of the macroinitiator 3, successful acylation
with 2-bromoisobutyryl bromide is evident from the disappearance of the O-H and
the formation of the ester linkage comes from the strong ester carbonyl peak at
1732 cm-1. The FT-IR spectrum of the homopolymer, 4a, had
characteristic peaks similar to those of 3, with the common features being (i)
absence of O-H, (ii) presence of CH2 asymmetric and CH2
symmetric frequencies, and (iii) >C=O around 1730 cm-1. Formation
of TEGA was also confirmed by FT-IR from the aliphatic CH2 peaks
around 2900 cm-1 and the ester carbonyl peak at 1722 cm-1.
1H-NMR of the
phthalonitrile precursor 1 in DMSO-d6
shows chemical shifts for the aromatic protons at δ=7.62(m), for the OH protons
at δ= 4.56(t), for the ArOCH2 protons at δ=4.28 as a triplet and for
all the other CH2O protons at δ=3.39-3.77 as multiplet. The chemical
shifts for these protons remain almost the same in the CuPc (2), with the aromatic protons at δ=7.4,
OH protons at δ=4.25, ArOCH2 protons at δ=3.91 and CH2O
protons at δ=3.6-3.8. In the 1H-NMR spectrum of the macroinitiator 3, the
presence of a singlet at δ=1.86, due to the chemical shift of the protons in
the CH3 groups at the end of the side chains, proves that the esterification
step has been successful.
The peaks for the ArOCH2 protons are at
δ=4.22 and for the CH2OCO protons at δ=3.75. Besides the aromatic
protons and the ArOCH2 protons at δ=7.26 and δ=4.22, respectively,
the 1H NMR spectrum of the poly-t-BA (4a) exhibits peaks at δ=2.2 for CH protons of the polymer backbone,
at δ=1.9 for the CH2 of the polymer backbone and at δ=1.43 (CH3)3C
protons of the t-BA ester group. The success of the copolymerization reactions
to synthesize 4b is conspicuous from
its 1H NMR spectra. It shows peaks attributed to the tert-butyl
resonance (δ= 1.44) as well as the to the OCH3 resonance (δ=3.38,
singlet). The 1H-NMR spectrum of TEGA monomer (5) shows three doublets of
doublet for the three protons in the C=C at δ=6.35, δ=6.1 and δ=5.79 and the
OCH3 resonance at δ=3.33.
The UV-Vis spectroscopy of compounds 2-4 was
performed in DCM. All exhibit the characteristic Q band of Pc molecules. Further
study was carried out with the homopolymer (4a) and the copolymer (4b)
in order to investigate their aggregation properties at low concentrations. This
was done by studying their UV-Vis absorption behavior at different
concentrations and plotting the intensity of their Q-bands at different
concentrations against the sample concentration at selected absorption
wavelength (corresponding to the wavelength of the Q bands). The linear
relationship between concentration and absorbance, as displayed in Figure 1 and Figure 2, strictly follows
the Lambert-Beer Law, suggesting that neither of them form aggregates at low
concentrations.
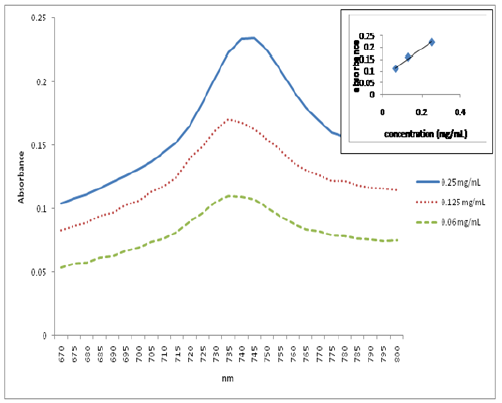
Figure 1: Plot of UV-Vis Absorption Spectra of Poly-t-BA (4a) at Different Concentrations against Wavelength in DCM. Inset: Plot of the Q Band Absorbance versus Concentration at 735 nm.
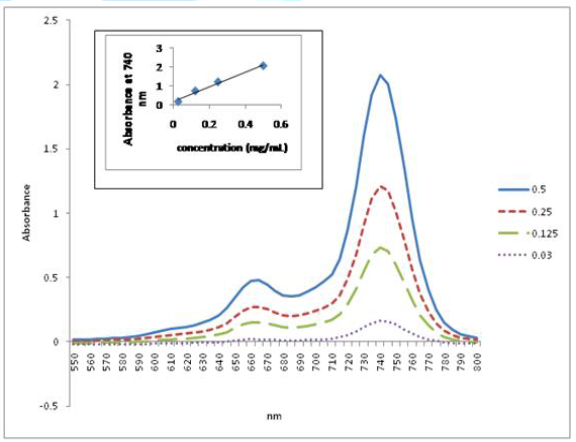
Figure 2: Plot of UV-Vis Absorption Spectra of Poly-t-BA-poly-TEGA (4b) at Different Concentrations against Wavelength in DCM. Inset: Plot of the Q Band Absorbance versus Concentration at 735 nm.
The solubility properties of compounds (1-4),
summarized in Table 1, indicating
that the copolymers are soluble in polar solvents.
Study
of Self-Assembly Properties
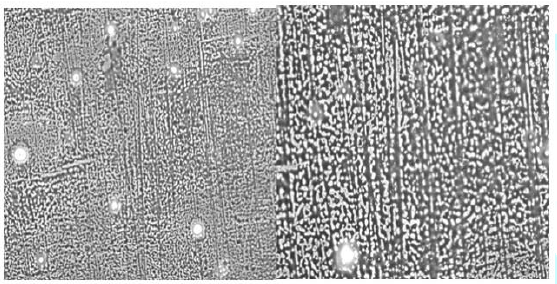
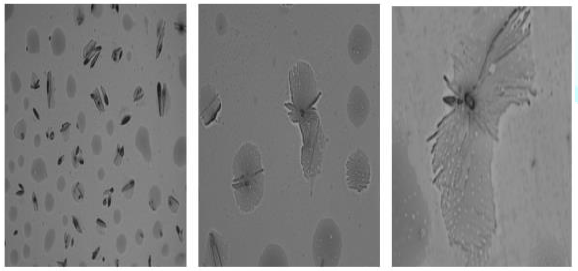
A preliminary study was carried out to investigate
the self-assembly properties of the CuPc-core block copolymer
poly-(t-BA)-poly-(TEGA) (4b), using optical microscopy, AFM and SEM. The copolymer
was dissolved in methanol (10 mg per mL). A drop of it was placed on the
substrate and the solvent was allowed to evaporate completely.
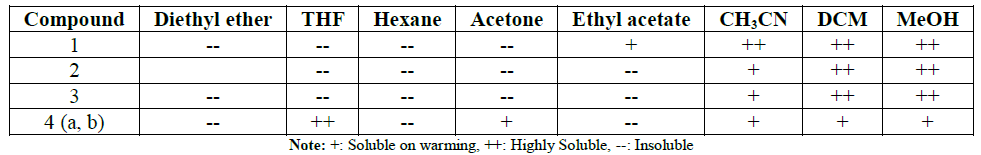
Table 1: SolubilityCharacteristics of Compounds 1-4.