Due to recent advances in computational
chemistry, not only computers and analysis programs in crystal structure
analysis, but also TD-DFT calculations related to intramolecular electronic
states and energies have been easily performed, and detailed discussions have
become possible. On the other hand, how to discuss information such as
intermolecular interactions, molecular assembly states, and packing, which is
beyond the scope of application of each computational chemistry program and
theory, is still a subject of research. Here, authors would like to consider
what is possible and difficult using a program, for example, Hirshfeld surface
analysis.
Introduction
With the aid of computational
chemistry, current coordination chemistry has developed to reach deep
understanding such as optimized structures, electronic structures (simulated
spectra), and reactivity based on thermodynamic
energy. In particular, DFT calculation provides detailed information of
metal complexes including organic ligands as well as metal ions. Besides
intramolecular information, however, coordination chemists may also be
interested in intermolecular effects of hybrid materials or photochemistry or
spectroscopy associated with spatially asymmetric conditions to discuss their
metal complexes. One of the ways to overcome such situations may be to employ
conventional physical frameworks based on the DFT results for molecular
information (Scheme 1), which was
proposed by us in certain international conference.
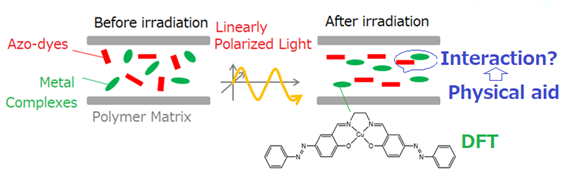
Scheme1:Concept of one of hybrid approaches of DFT and physical principle for dipole-dipole interaction..
In recent years, calculations for crystal structure
analysis of small molecule compounds have generally been performed relatively
smoothly by well-developed programs, from various data processing programs to theoretical
calculations for discussion using results. In fact, in our laboratory, we
examined whether it is possible to discuss the coordination bond of metal
complexes by comparing the electron density by experiment with the electron
density by TD-DFT
theoretical calculation using a conventional program [1].
Results
and Discussion
With the ultimate goal of expressing the
antibacterial activity of the novel hydrazone ligand complex, we are conducting
docking simulations with proteins based on crystal structure analysis data, and
are studying intermolecular interactions between ligands and proteins. Focusing
on molecular recognition by ligands outside the complex molecule, crystal
structure analysis (re-determination) of Nicotinohydrazide derivative hydrazone
with only a ligand having a new structure and intermolecular mutual as crystals
by Hirshfeld
surface analysis compared with similar compounds containing different
solvents (Figure 1) or isomers at
the pyridine site [2-6] (Figure 2).
In order to
visualize the intermolecular interactions in the crystal of the compounds, a
Hirshfeld surface analysis was performed with a Crystal Explorer 17.5 program.
The fingerprint plot for this structure shows typical wings. The percentage contribution
of each intermolecular interaction to the Hirshfeld surface area is shown in
the figures. Hirshfeld surface analysis is a computational
method that discusses intermolecular interactions in the case of a single
compound that forms a molecular crystal. Except for the method of forming
molecular crystals, that is, the conformation that is the most stable in terms
of energy and the prediction of crystal packing. It must be still a difficult
problem.
Various ligand-protein docking calculation programs
are known for intermolecular interactions between small molecule compounds and
other substances. The basis is the crystal structure of proteins and ligands
and the DFT-optimized structure of ligands. It has the meaning of simplifying
complicated factors in order to evaluate the contribution of shape and various
interactions as one numerical value called "docking score". On the
other hand, similar discussions are difficult for organic-inorganic
nanocomposites whose three-dimensional structure is undecided. For example, is
it possible to use the description of curved surfaces using mathematics of
differential geometry to express only the shape?
Furthermore, is it possible to discuss the hydrophobic/hydrophilic
interaction with a solvent (around the dielectric environment) in solution
by diverting the donor or acceptor of the potential interaction by Hirshfeld
surface analysis? Should ordinary physical laws be used in addition to the
knowledge of computational chemistry to consider interactions with surfactants
and solvent micro-spaces (nano-droplets, etc.)?
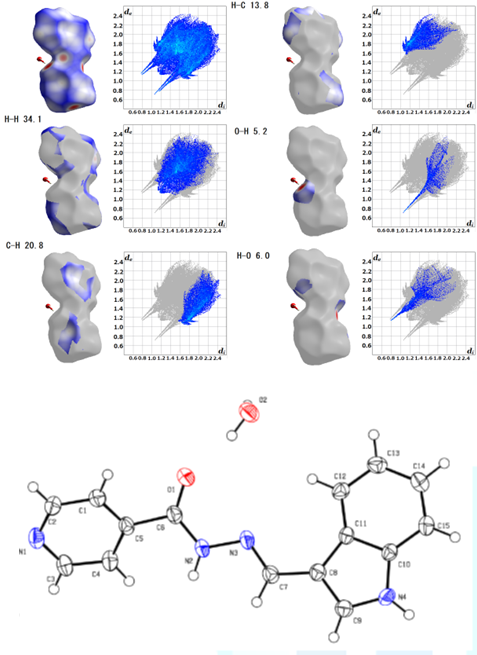
Figure1:Hirshfeld surface analysis of C15H12N4O.H2O[2].
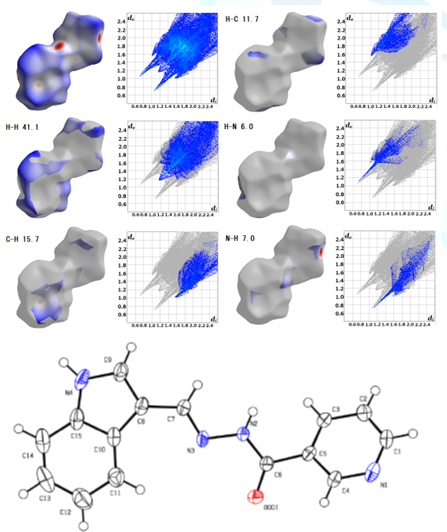
Figure2:Hirshfeld surface analysis and ORTEP drawing of C15H12N4O[6].
Conclusion
While simple analytical calculations and
computational theoretical chemistry have become widespread, it is another
matter whether or not to discuss all substances of interest today. Of course,
high-precision computational chemistry has a range of applications. It may be
less accurate, but humans may apply the laws of physics (using computational
chemistry data) to consider it.
Acknowledgement
This work was supported by a Grant-in-Aid for
Scientific Research (A) KAKENHI (20H00336). The authors thank Prof. Emmanuel N.
N for at Department of Chemistry, University of Buea for suggestion of
hydrazone compounds.
References
- Takiguchi Y, Onami Y, Haraguchi T and Akitsu T. Crystallographic
and computational electron density of dx2-y2 orbitals of
azo-Schiff base metal complexes using conventional programs (2021) Molecule 26:
551. https://doi.org/10.3390/molecules26030551
- Xia LY, Wang WL, Wang SH, Huang YL and Shan S. N'-[(E)-3-Indol-3-ylmethyl-ene]isonicotino-hydrazide
monohydrate (2009) Acta Cryst E 65: o1900-o1900. https://doi.org/10.1107/s1600536809027329
- Jing ZL, Cheng WW, Chen X and Ming Y.
2′-(1H-Indol-3-ylmethylene)isonicotinohydrazide ethanol solvate (2006) Acta
Cryst E 62: o1360-o1361. https://doi.org/10.1107/S1600536806008361
- Saranya S, Haribabu J, Bhuvanesh NSP, Karvembu R and
Gayathri D. Crystal structures of the Schiff base derivatives
(E)-N′-[(1H-indol-3-yl)methylidene]isonicotinohydrazide ethanol monosolvate
and
(E)-N-methyl-2-[1-(2-oxo-2H-chromen-3-yl)ethylidene]hydrazinecarbothioamide
(2017) Acta Cryst E 73: 594-597. https://doi.org/10.1107/S205698901700411X
- Tai XS, Yin XH, Tan MY and Li YZ. 3-Indolylformaldehyde
isonicotinoylhydrazone methanol solvate (2003) Acta Cryst E 59: o681-o682. https://doi.org/10.1107/S1600536803008286
- Gwaram
NS reported as CCDC 882156, CSD ABARAZ (2016), unpublished as a paper.
Corresponding
author
Takashiro Akitsu, Department of
Chemistry, Faculty of Science, Tokyo University of Science, 1-3 Kagurazaka,
Shinjuku-ku, Tokyo 162-8601, Japan. E-mail: akitsu2@rs.tus.ac.jp
Citation
Akitsu T, Suda S and Katsuumi N. Beyond
the scope of each computational chemistry (2021) Edelweiss Chem Sci J 4: 25-26.
Crystal structure
analysis, Hirshfeld surface analysis, TD-DFT, Intermolecular interaction, Hybrid
materials


