

Research Article :
A
theoretical calculation of the fully optimized geometries and electronic
structures of the metal-free Tetra-2,3-Pyridino-Porphyrazine (TPdPzH2*),
N,N-Dideuterio (TPdPzD2*), and Magnesium (TPdPzMg*)
tetra-3,4-pyridino-porphyrazine has been conducted with the density functional
B3LYP level using the 6-31G(d) basis set. A comparison among the different
Phthalocyanine (Pc) derivatives, including Tetra-2,3-Pyridino-Porphyrazine
(TPdPzH2) compounds, for the geometry, molecular orbital, and atomic
charge was made. The substitution effect of the N atoms and the isotopic effect
of D atoms on the properties of these compounds were discussed. The farther the
heterocyclic N atoms in the benzo rings from 16-membered ring are, the smaller
it influence the size of the central hole, the bond lengths and bond angles of
16-membered ring, the HOMO-LUMO gaps, and the atomic charges on the core Pz
fragment. In other words, the properties of TPdPz* compounds are
closer to Pc than TPdPz. With the order of H2Pc Phthalocyanines (Pcs) have been
widely used as organic dyestuffs since their first synthesis early last century
because of their intense absorption of light in the visible and ultra-violet
regions and their high chemical and thermal stabilities [1,2]. They have also
attracted considerable interest due to their applications in modern science and
technology on the basis of their special electric and photoelectric properties: semiconductivity,
photoconductivity and luminescence [3]. The
benzo rings of Pc compounds can be substituted by heterocycles such as
thiophene, thionaphthalene, pyridine, or pyrazine, thus forming heterocyclic Pc
analogues. The aza-Analogues of the Phthalocyanines (aza-Pcs) have been
extensively studied over the past three decades. Potential applications include
their use as textile bleaching agents, photoinactivators for controlling growth
of microorganisms, catalysts for oxygen reduction, materials for eletrochromic
displays, and media for optical data storage with large memory capacity,
inhibitor of thermal degradation of polymers and photosensitizers for photodynamic therapy of cancer [4-9]. The
synthesis of heterocyclic Pc analogues was initially reported in 1937 by
Linstead and co-workers [10,11]. A significant contribution to the development
of the aza-Pcs chemistry was made by Galpern and Lukyanets [12] who described
the synthesis and properties of a series of aza-Pcs. Previously, Density Functional Theory (DFT) methods
have been extensively employed investigating Pcs and their derivatives by Zhang
et al [13-18]. We have also approached the molecular, electronic structures and
vibrational spectra for metal-free (TPdPzH2), N,N-Dideuterio (TPdPzD2)
and Magnesium (TPdPzMg) tetra-2,3-pyridino-porphyrazines and metal-free (TPyPzH2),
N,N-Dideuterio (TPyPzD2) and Magnesium (TPyPzMg) tetra-2,3-pyrazino-porphyrazines [19,20]. Moreover,
theoretical investigation of the molecular, electronic structures and
vibrational spectra of a series of transition metal phthalocyanines and
naphthalocyanine have been performed using the density functional theory, and
ideal results have been achieved [21,23]. All the previous research works
demonstrated that the density functional B3LYP method performs well in the
calculation for phthalocyanines
and their analogues,
thus it should also be appropriate for the tetra-3,4-pyridino-porphyrazine
complexes. Computer
simulation as an attractive alternative to experiment can provide valuable
information. So, to get more insights into the molecular and electronic
structures, it is worthwhile to conduct quantum chemistry calculations. In the
present paper, we report theoretical calculations of molecular, electronic
structures of metal-free Tetra-3,4-Pyridino-Porphyrazine (TPdPzH2*),
its N,N-Dideuterio Derivative (TPdPzD2*) and Magnesium
complex (TPdPzMg*). The substitution effect of the N atoms and the
isotopic effect of D atoms on these properties of these compounds were
discussed. Some interesting and meaningful results were obtained. The
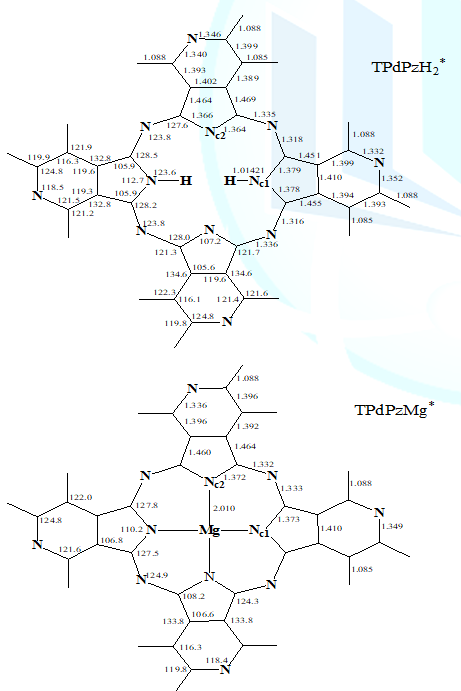
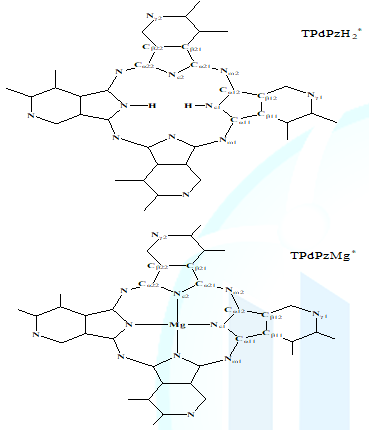
structures of the tetra-3,4-pyridino-porphyrazine are shown in Figure 1. The 6-31G(d) basis set was
used at the density functional B3LYP level for geometry optimization and
calculations. The default Mulliken method was used for atomic charge
calculation. The Berny algorithm using redundant
internal coordinates was employed in energy minimization and tight convergence
criteria were used throughout [24]. C4h symmetry for TPdPzMg*
and C2h for TPdPzH2* and TPdPzD2*
in the input structures were detected and then enforced by the program. Using
the energy-minimized structures generated in the previous step, normal
coordinate analyses were carried out. All calculations were carried out using
Gaussian09 program. Molecular
structures At
density functional B3LYP level, the energy-minimized structure of TPdPzMg*
calculated has C4h symmetry, and both TPdPzH2*
and TPdPzD2* have C2h symmetry. No imaginary
vibration is predicted in the frequency calculations, indicating that the
energy-minimized structures are true energy minima. Corresponding structural
parameters are illustrated in Figure 1. Whats interesting is that N,N-Dideuterio-Derivative (TPdPzD2*)
cannot change the structural parameters in metal-free-derivative (TPdPzH2*),
thus only the bond lengths and bond angles of TPdPzH2* herein
are discussed. Considering the substitution effect of the N atoms on the molecular
structure, except that N-H bond lengths maintain six significant digits to
compare, the other structure parameters retain four. As
shown in Figure 1, the N-H bond length is 1.01421 Å for TPdPzH2*,
which is slightly longer than that of TPdPzH2 (1.01418 Å), and
longer than that of H2Pc (1.01399 Å) [24]. These indicate that the
N-H bond strengths of the three molecules vary with the sequentially decreasing
order of H2Pc>TPdPzH2>TPdPzH2*,
which also implies that the acidity of the inner N-H bonds of the three
molecules sequentially increase with the trend of H2Pc Analysis
of the structure parameters of TPdPzMg* shows that the average size
of the central hole (Nc-Nc distance) is 4.021 Å, which is larger than those of
MgPc (4.017 Å), but smaller than that of TPdPzMg (4.024 Å) [19]. This indicates
that the size of the central hole increases with the order of MgPc Electronic
structures and molecular orbitals The
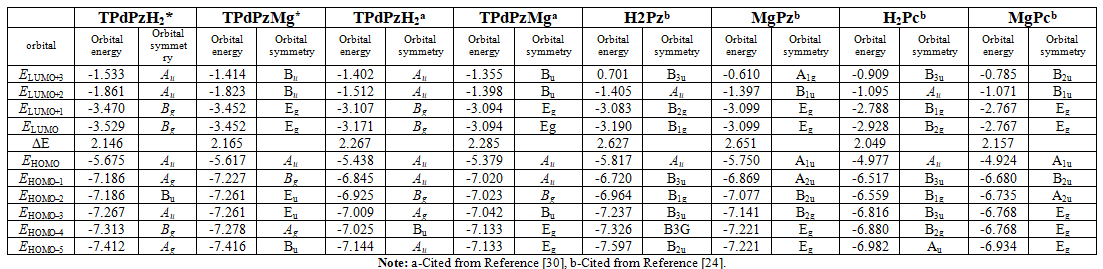
energies of the molecular orbitals from HOMO-5 to LUMO+3 of TPdPzH2*
and TPdPzMg* are comparatively listed in Table 1 and shown in Figure 2.
Herein, HOMO and LUMO represent the highest occupied molecular orbital and the
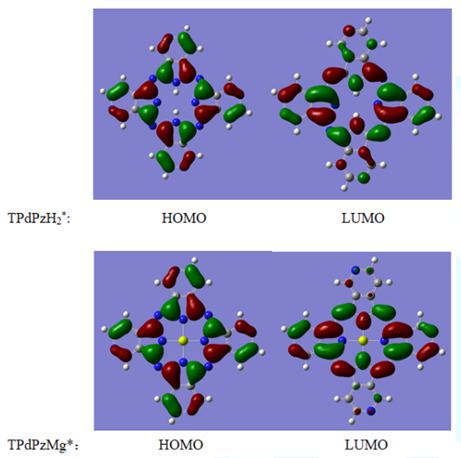
lowest unoccupied molecular orbital, respectively. Figure 3 shows the scheme of HOMO and LUMO of TPdPzH2*
and TPdPzMg*. It is worth noting that N, N-dideuterio-derivative
(TPdPzD2*) cannot change the molecular orbital energy
levels in metal-free-derivative (TPdPzH2*). In this
paper, the energies of TPdPzH2* are only discussed. Figure 3: Graphic
representation of HOMO and LUMO of TPdPzH2* and TPdPzMg*. There
are two characteristic absorption bands (the Q band and Soret or B band) in UV-visible spectra of
porphyrazines
and their analogues with fused heterocycles. The Q band in the visible region
originates from the HOMO→LUMO electronic transition [26]. In practical
application, people pay attention to Q band. Due
to the extension of π-conjugation, the HOMO-LUMO gaps of TPdPzH2*
and TPdPzMg* (2.146 and 2.165eV) are smaller than those of H2Pz
and MgPz (2.627 and 2.651 eV), indicating that the Q bands of TPdPzH2*
and TPdPzMg* shift to a longer wavelength side than those of H2Pz
and MgPz [13]. According to Linstead, some Pz derivatives show Q band peaks at
535-617 nm, while the TPdPz* compounds at 574-672 nm [27,28]. These
data agree very well with the above-mentioned calculation results. The
HOMO-LUMO gaps of TPdPzH2*
and TPdPzMg* (2.146 and 2.165 eV) are found to be a bit larger than
those of H2Pc and MgPc (2.049 and 2.157 eV), and smaller than those
of TPdPzH2 and TPdPzMg (2.266 and 2.285 eV) [13,19]. This
straightens out the hypsochromic shift of the Q band in the electronic spectra
of these aza phthalocyanine analogues, on the other hand, interprets electronic
spectra in the visible region of TPdPz* compounds are identical to
the spectra of the corresponding Pcs due to the HOMO-LUMO gaps of them are near
each other [27,29]. According to Galpern, the substitution of the CH groups in
the Pc benzo rings adjacent to the porphyrazine macrocycle with N atoms causes
a remarkable hypsochromic shift in the visible part of the electronic
absorption spectrum. The magnitude of this shift increases from tetra-aza-Pcs
(20-25 nm, 550-780 cm-1) to the octa-aza derivatives (40-60 nm,
870-1260 cm-1) depending on the nature of the central metal atom
[30,31]. The
energy levels of the HOMO-5 to LUMO+3 orbitals for TPdPz* compounds
all decrease compared with those of the corresponding Pc and TPdPz compounds.
Moreover, the magnitude of all the energy levels is in the order of TPdPz* Compared
with H2Pc and MgPc, the reduction of HOMO of TPdPzH2*
and TPdPzMg* are 0.698 and 0.693 eV, while the reduction of LUMO are
0.601 and 0.685 eV, respectively, and the decrease of HOMO are slightly larger
than those of LUMO [13]. This is due to the presence of the p-deficient
annulated pyridine rings in the TPdPz* unit as compared with the
isoelectronic benzene rings in the Pc unit. The lowering of HOMO for TPdPzH2*
and TPdPzMg* compared with those of TPdPzH2 and TPdPzMg
are 0.238 and 0.238 eV, which are smaller than those of LUMO, 0.358 and 0.358
eV, respectively [19]. The possible reason is that the heterocyclic N atoms on
TPdPz* compounds are farther from the central 16-membered ring than
those on TPdPz compounds, and they affect the energy levels of the LUMO
orbitals are larger than those of the HOMO ones. This interprets the HOMO-LUMO
gaps of TPdPzH2* and TPdPzMg* are smaller than
those of TPdPzH2 and TPdPzMg. Atomic
charges Both
phthalocyanine and aza-Pcs compounds contain basic porphyrazine structural
units and additional p-conjugated rings. This sequential
increase in the number of p-conjugated
aromatic rings
gives rise to increased polarization of basic porphyrazine ring, which causes
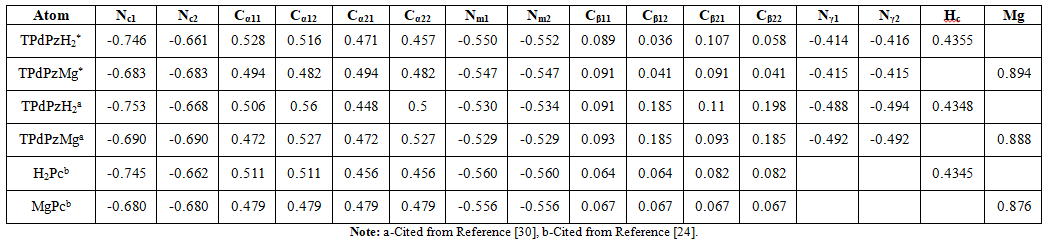
the change of p-electron energy. The atomic charge of
the core Pz fragment and the external N atoms for TPdPzH2*
and TPdPzMg* compounds are comparatively listed in Table 2. It is worth noting that
N,N-dideuterio-derivative (TPdPzD2*) cannot change the
atomic charge in metal-free-derivative (TPdPzH2*), thus
the atomic charge of TPdPzD2* herein is not discussed. In
order to show the difference between TPdPzH2* and TPdPzH2,
the significant digits of the atomic charges are maintained
three except that four significant digits retained by Hc. The atom symbols for
TPdPzH2* and TPdPzMg* are shown in Figure 4. Compared
with the TPdPz compounds, the charges of the central Nc for the TPdPz*
compounds become less negative, while the meso-N atoms (Nm) become
more negative, which means that the charges of these atoms for TPdPz* compounds
are nearer to those of Pc compounds [19]. Table 2: Atomic charge
(in e) for TPdPzH2* and TPdPzMg*. In
addition, all the other charges of the corresponding atoms in the core Pz
fragment for TPdPz* compounds are nearer to those of Pc compounds. This implies
that the farther the heterocyclic N atoms in the benzo rings from the central
16-membered ring are, the smaller they influence the charges of the atoms on
the core Pz fragment. That is to say, the atom charges of the core Pz fragment
in TPdPz* compounds are nearer to those of Pc, which basically agree
with the experimental electronic spectra in the visible region of TPdPz*
compounds being identical to the spectra of the corresponding Pcs [29]. The
atomic charge of the central Hc (0.4355 e) of TPdPzH2* is
larger than that of TPdPzH2 (0.4348 e) and H2Pc (0.4345
e), which is consistent with the acidity of the inner N-H bonds of the three
molecules (H2Pc According
to our calculation, we found that the farther the heterocyclic N atoms in the
benzo rings from 16-membered ring are, the smaller it influence the size of the
central hole, the bond lengths and bond angles of 16–membered ring, the HOMO-LUMO
gaps, and the charges of the atoms on the core Pz fragment. That is to say,
TPdPz* compounds are nearer to Pc compounds than TPdPz in structure,
energies of the molecular orbitals, and atomic charges. The acidity of the
inner N-H bonds of the three molecules sequentially increase with the order of
H2Pc The
authors thank the National Natural Science Foundation of China (Grant No.
20501011). 1.
Leznoff
CC and Lever ABP (Eds). Phthalocyanines: properties and applications (1989) to
(1996) VCH Publishers, United States. 2.
McKeown
NB. Phthalocyanine materials: synthesis, structure and function (1998)
Cambridge University Press, United States. 3.
Kadish
KM, Smith KM and Guilard R (Eds). The porphyrin handbook (2000) Academic Press,
United States, pp-1-10. 4.
Tanaka
AA, Fierro C, Sharson DA and Yaeger E. Oxygen reduction on adsorbed iron
tetrapyridinoporphyrazine (1989) Mater Chem Phys 22: 431. 5.
Yamano
M, Kashivazaki N, Yamamoto M and Nakano T. A storage type electrochromic
display utilizing Poly-Co-Qtpp films (1987) Jpn J Appl Phys 26: L1113. https://doi.org/10.1143/JJAP.26.L1113 6.
Schlettwein
D, Woehrle D and Jaeger NI. Reversible reduction and reoxidation of thin films
of tetrapyrazinotetraazaporphyrines (1989) J Electrochem Soc 136: 2882-2886. 7.
Kasuga
K, Nishikori K, Mihara T, Manda M, Sogabe K, et al. Preparation of bis (tetra-2,3
pyradinoporphyrazinato) ytterbium(III) (1990) Inorg Chim Acta 174: 153-154. https://doi.org/10.1016/S0020-1693(00)80292-3 8.
Teuchner
K, Pfarrherr A, Stiel H, Freyer W and Leupold D. Spectroscopic properties of
potential sensitizers for new photodynamic therapy mechanisms via two-step
excited electronic states (1993) Photochem Photobiol 57: 465-471. https://doi.org/10.1111/j.1751-1097.1993.tb02320.x 9.
Kudrevich
SV, Galpern MG and van Lier JE. Synthesis of octacarboxytetra (2,3-pyrazino) porphyrazine:
Novel water soluble photosensitizers for photodynamic therapy (1994) Synthesis
779-781. https://doi.org/10.1055/s-1994-25571 10.
Linstead
RP, Noble EG and Wright JM. Phthalocyanines. Part IX. Derivatives of thiophen,
thionaphthen, pyridine, and pyrazine, and a note on the nomenclature (1937) J
Chem Soc 911-921. https://doi.org/10.1039/jr9370000911 11.
Anderson
JS, Bradbrook EF, Cook AH and Linstead RP. Phthalocyanines associated
compounds. Part XIII. Absorption spectra (1938) J Chem Soc 1151-1156. https://doi.org/10.1039/jr9380001151 12.
Kudrevich
SV and van Lier JE. Azaanalogs of phthalocyanine: syntheses and properties
(1996) Coordin Chem Rev 156: 163-182. https://doi.org/10.1016/s0010-8545(96)01251-9 13.
Zhang
XX, Zhang YX and Jiang JZ. Geometry and electronic structure of metal free
porphyrazine, phthalocyanine and naphthalocyanine as well as their magnesium
complexes (2004) J Mol Struct Theochem 673: 103-108. https://doi.org/10.1016/j.theochem.2003.12.004 14.
Ma
RM, Guo P, Yang LL, Guo LS, Zhang XX, et al. Theoretical screening of -NH2-,
-OH-, -CH3-, -F-, and -SH-substituted porphyrins as sensitizer candidates for
dye-sensitized solar cells (2010) J Phys Chem A 114: 1973-1979. https://doi.org/10.1021/jp909787t 15.
Yang
LL, Guo LS, Chen QQ, Sun HF, Yan H, et al. Substituent effects on zinc
phthalocyanine derivatives: A theoretical calculation and screening of
sensitizer candidates for dye-sensitized solar cells (2012) J Mol Graph Model
38: 82-89. https://doi.org/10.1016/j.jmgm.2012.08.006 16.
Yang
LL, Guo LS, Chen QQ, Sun HF, Liu J, et al. Theoretical design and screening of
panchromatic phthalocyanine sensitizers derived from TT1 for dye-sensitized
solar cells (2012) J Mol Graph Model 34: 1-9. https://doi.org/10.1016/j.jmgm.2011.12.001 17.
Zhang
XX, Du YC, Chen QQ, Sun HF, Pan TT, et al. Theoretical screening of novel
alkyne bridged zinc porphyrins as sensitizer candidates for dye-sensitized
solar cells (2014) Spectrochim Acta Part A 133: 514-520. https://doi.org/10.1016/j.saa.2014.06.015 18.
Zhang
XX, Chen QQ, Sun HF, Pan TT, Hu GQ, et al. Theoretical design and screening of
alkyne bridged triphenyl zinc porphyrins as sensitizer candidates for
dye-sensitized solar cells (2014) Spectrochim Acta Part A 118: 564-571. https://doi.org/10.1016/j.saa.2014.06.015 19.
Liu
ZQ, Zhang XX, Zhang YX, Li RJ and Jiang JZ. The molecular, electronic
structures and vibrational spectra of metal-free, N,N′-dideuterio and
magnesiumtetra-2,3-pyridino-porphyrazines: Density functional calculations
(2006) Spectrochim Acta Part A 65: 467-480. https://doi.org/10.1016/j.saa.2005.11.027 20.
Liu
ZQ, Zhang XX, Zhang YX and Jiang JZ. The molecular, electronic structures and
IR and Raman spectra of metal-free, N,N-dideuterio, and magnesium
tetra-2,3-pyrazino-porphyrazines: Density functional calculations (2007) Vib
Spectro 43: 447-459. https://doi.org/10.1016/j.vibspec.2006.06.002 21.
Liu
ZQ, Zhang XX, Zhang YX and Jiang JZ. Theoretical investigation of the
molecular, electronic structures and vibrational spectra of a series of first
transition metal phthalocyanines (2007) Spectrochim Acta Part A 67: 1232-1246.
https://doi.org/10.1016/j.saa.2006.10.013 22.
Liu
ZQ, Chen Z-X, Jin BB and Zhang XX. Theoretical studies on the structures and
vibrational spectra of Ni, Pd, and Pt phthalocyanines (2011) Vib Spectro 56:
210-218. https://doi.org/10.1016/j.vibspec.2011.02.010 23.
Liu
ZQ, Chen Z-X and Jin BB. Density functional theory studies on the structures
and vibrational spectroscopic characteristics of nickel, copper and zinc naphthalocyanines
(2019) Spectrochim Acta Part A 217: 8-17. https://doi.org/10.1016/j.saa.2019.03.029 24.
Peng C, Ayala PY, Schlegel HB and Frisch MJ. Using
redundant internal coordinates to optimize equilibrium geometries and
transition states (1996) J Comp Chem 17: 49-56. https://doi.org/10.1002/(sici)1096-987x(19960115)17:1%3C49::aid-jcc5%3E3.0.co;2-0 25.
Woehrle
D, Gitzel J, Okuro I and Aono S. Photoredox properties of
tetra-2,3-pyridinoporphyrazines (29H,31H-tetrapyrido[2,3-b: 2′, 3′-g :
2″,3″-l:2‴,3‴-q]porphyrazine) (1985) J Chem Soc Perkin Trans 2: 1172. https://doi.org/10.1039/P29850001171 26.
Hamdoush
M, Ivanova SS, Koifman OI, Koskina M., Pakhomov GL, et al. Synthesis, spectral
and electrochemical study of perchlorinated tetrapyrazinoporphyrazine and its
AlIII, GaIII and InIII complexes (2016) Inorg Chim Acta 444: 81-86. https://doi.org/10.1016/j.ica.2016.01.029 27.
Linstead
RP and Whalley M. Conjugated macrocycles Part XXII Tetrazaporphin and its
metallic derivatives (1952) J Chem Soc 4839-4846. https://doi.org/10.1039/jr9520004839 28.
Dreich SV and van Lier JE. Azaanalogs of
phthalocyanine: syntheses and properties (1996) Coordin Chem Rev 156: 163-182. https://doi.org/10.1016/s0010-8545(96)01251-9 29.
Yokote
M, Shibamiya F and Yokomizo H. Nickel-Phthalocyanine-N-Isolog (Nickel-tetra-2,
3Pyridinoporphyrazine and Nickel-tetra-3, 4Pyridinoporphyrazine) (1969) J Synth
Org Chem Jpn 27: 340. 30.
Galpern
EG, Lukyanets EA and Galpern MG. Effect of aza-substitution on electron
absorption-spectra of phthalocyanines (1973) Izv Akad Nauk SSSR Ser Khim 9. 31.
Galpern
MG and Lukyanets EA. Phthalocyanines and related compounds. 7. tetra-2, 3-benzo
[G] quinoxalinoporphyrazines (1971) Zh Obsch Khim 41: 2549. Zhongqiang Liu, Associate Professor, Research Center
of Theoretical and Computational Chemistry, School of Chemistry and Chemical
Engineering, Qilu Normal University, Jinan 250200, PR China, Tel: +86 531
66778025, E-mail: warlter2000@163.com Liu Z and Zhang X. Density functional calculations
on the molecular, electronic structures of metal-free, N,N-dideuterio and
magnesium tetra-3,4-pyridino-porphyrazines (2019) Edelweiss Chem Sci J 2:
40-44. Tetra-3,4-pyridino-porphyrazine, DFT method, Molecular
structure, Electronic structure, Phthalocyanines.Density Functional Calculations on the Molecular, Electronic Structures of Metal-Free, N,N-Dideuterio and Magnesium Tetra-3,4-Pyridino-Porphyrazines
Zhongqiang Liu and Xianxi Zhang
Abstract
Full-Text
Introduction
Computational
Method
Results
and Discussion

Conclusions
Acknowledgements
References
Citation
Keywords
