Introduction
Dopamine
(DA) dysfunction [1,2], glutamate dysfunction [3,4], neurodevelopmental deficits [5,6], or neural stem cell (NSC)
dysfunction [7,8] are well-known hypotheses for etiology of schizophrenia. DA
dysfunction hypothesis suggested that mesolimbic DA hyperactivity caused
positive symptoms such as paranoid-hallucinatory state of schizophrenia [1,2].
It is also explained by the efficacy of DA D2 blockers for paranoid-hallucinatory
state and also by hallucinogenic acts of DA stimulants including
methamphetamine or amphetamine [1,2]. Glutamate dysfunction theory was induced
by the fact that intake of phencyclidine (PCP), an antagonist of N-methyl-D-aspartate
(NMDA) receptor, produces equivalent to negative symptoms of schizophrenia,
such as withdrawal or flattened affect, as well as positive symptoms [3,4]. The
neurodevelopmental deficits hypothesis implicates that schizophrenia is the
consequence of prenatal abnormalities resulting from the interaction of genetic
and environmental factors [5,6]. NSC dysfunction has also been shown to be a
cause of schizophrenia
[7,8]. Although mesolimbic DA hyperactivity [1,2] has been well documented in
pathogenesis of schizophrenia, the molecular basis of this mechanism has not
yet been detailed. In the present article, the author shows the rational of the
reduction of putative trace amine (TA)-producing neurons (D-neurons), that is,
ligand neurons of TA-associated receptor, type 1 (TAAR1), in the striatum in the
pathogenesis of mesolimbic DA hyperactivity of schizophrenia [9]. The novel hypothesis,
“D-cell hypothesis of schizophrenia”, is a critical theory to link NSC dysfunction
hypothesis with DA hypothesis in etiology of schizophrenia.
D-neuron
(trace amine neuron) The
“D-cell” was described, by Jaeger et al. [10], in 1983 in the rat central
nervous system and was defined “the non-monoaminergic aromatic L-amino acid
decarboxylase (AADC)-containing cell” [10]. AADC is an equivalent enzyme to
dopa decarboxylase (DDC). The D-cell contains AADC but not dopaminergic nor
serotonergic [10]. Then, it is natural that the D-cell is thought to produce
TAs [11,12], such as β-phenylethylamine (PEA), tyramine, tryptamine
and/or octopamine. AADC is the rate-limiting enzyme for TA synthesis. However, it is
confusing that these TAs are also “monoamines”, as each one has one amino residue.
It would be better to use the nomenclature of “TA cells” for D-cells, and “TA
neurons” for D-neurons. In the present article, the author uses the words,
D-cell and D-neuron, signifying TA cell and TA neuron, respectively. The
localizations of D-cells were specified into 14 groups, from D1 (the spinal
cord) to D14 (the bed nucleus of stria terminalis) in caudo-rostral order of
the rat central nervous system using AADC immunohistochemistry [13]. In this
usage, the classification term “D” means decarboxylation. In rodents [14,15], a
small number of D-cells in the striatum were rostrally described and confirmed
to be neurons by electronmicroscopic observation [14,15]. I reported in 1997, “dopadecarboxylating
neurons specific to the human striatum [16-19]”, that is, “D-neurons” in the
human striatum [18,20] (classified to be D15) [18], though monkey striatum did
not contain D-neurons [18]. In 2003, by using pathological and legal autopsy
brains of patients with schizophrenia, reduction of D-neurons in the striatum,
including nucleus accumbens (Acc) (classified to be D16) of patients with
schizophrenia [9,20] was also shown.
Trace
amine (TA)-associated receptor, type 1 (TAAR1)
C loning of TA
receptors in 2001 [21,22], elicited enormous efforts for exploring signal
transduction of these G-protein coupled receptors whose genes are located on
chromosome focus 6q23.1 [23]. The receptors have been shown to co-localize with
DA or adrenaline transporters in monoamine neurons and to modulate the
functions of monoamines [24-26]. The TAAR1 having a large number of ligands,
including, PEA, tyramine, 3-iodothyronamine, 3-methoxytyramine, normetanephrine
and psychostimulants, for example methamphetamine
3,4-methylenedioxymethamphetamine (MDMA) and lysergic acid diethylamide (LSD)
[21,23,26], has become a target receptor for exploring novel neuroleptics
[27,28]. However, endogenous TAAR1 ligands in the human central nervous system
have not yet been specified. TAAR1 knockout mice showed schizophrenia-like
behaviors with a deficit in prepulse inhibition [29,30]. TAAR1 knockout mice
showed greater locomotor response to amphetamine and released more DA (and noradrenaline)
in response to amphetamine than wild type mice [29]. It has been shown that
TAAR1 has a thermoregulatory function [30]. As is the important fact, it was
clarified that increased stimulation of TAAR1 receptors on cell membranes of DA
neurons
in the midbrain ventral tegmental area (VTA) reduced firing frequency of VTA DA
neurons [27-30]. This made the author to suspect the existence of critical role
of TAAR1 stimulation decrease for mesolimbic DA hyperactivity in schizophrenia.
“D-cell hypothesis” of schizophrenia
A theory, “D-cell
hypothesis”, for explaining mesolimbic DA hyperactivity in pathogenesis of
schizophrenia is shown in Figure 1. In brains of patients with schizophrenia,
dysfunction of NSC in the subventricular zone of lateral ventricle causes
D-neuron decrease in the striatum and Acc [8,31]. This induces TA decrease in
these nuclei, though direct evidences have not yet been demonstrated.
Enlargement of the lateral ventricle [32,33], a usual finding documented
in brain imaging studies of schizophrenia,
is probably due to NSC dysfunction in the subventricular zone [7,8]. The reduction
of TAAR1 stimulation on DA terminals of VTA DA neurons, caused by TA decrease,
would increase firing frequency of VTA DA neurons [28,30,31]. This increases DA
release and DA turnover in the Acc [2], resulting in mesolimbic DA hyperactivity.
It has been shown that D2 stimulation of NSC in the striatum inhibited
forebrain NSC proliferation [31,34]. Striatal DA hyperactivity may accelerate
D-neuron decrease, which accelerates hyperactivity of mesolimbic DA system.
Actions of D2 blocking agents in pharmacotherapy of schizophrenia might be
explained by blocking the inhibition to forebrain NSC proliferations, and also
by formation of TAAR1 ligands, such as 3-methoxytyramine and normetanephrine
[35]. It is consistent with clinical evidences that initial pharmacotherapy
using D2 antagonists is proved to be critical for preventing progressive
pathognomonic procedures of schizophrenia [36].
Disease
progression of schizophrenia and therapeutic strategies
D-cell
hypothesis not only links DA hypothesis with NSC dysfunction hypothesis, but
also explains the mechanisms of disease progression of schizophrenia as shown
in figure 1. To inhibit this cycle of pathological progression, intervention
indicated by ①~③, shown in Figure 1, is supposed to be effective.
Early studies have shown formation of
some TAAR1 ligands by administration of
D2 antagonists including haloperidol and chlorpromazine [35]. In recent animal
studies, effectiveness of TAAR1 ligands for schizophrenia-like symptoms of
schizophrenia model animals has been shown [28]. 2) D2
antagonists (Figure 1 ②) Duration of
untreated psychosis is a predictor of long-term outcome of schizophrenia.
Importance of early intervention for first episode schizophrenia
by using D2 antagonist has been emphasized. Chronic D2 blocker administration
has preventive effect for recurrence of psychoses. D2 antagonists may block
disease progression as shown in Figure 1. D2 antagonists have dual actions for
inhibiting this cycle of disease progression by also forming some TAAR1 ligands
(3-methoxytyramine, normetanephrine) which may increase TAAR1 stimulation as
shown in Figure 1 [35]. 3) Neurotrophic substances (Figure 1 ③) Disease progression would be inhibited by neurotrophic substances,
for example, brain-derived neurotrophic factor (BDNF), lithium,
anticonvulsants, or antidepressants.
These substances, having neurotrophic effects, activate NSC functions [37], and
inhibit striato-accumbal D-neuron decrease. 4) Intranasal administration of
drugs, expecting retrograde transport of neuroactive substances or their
precursors through the olfactory bulb, might be a novel therapeutic strategy (①~③). It is a possible preferable method of
administration, as it devoid of gastrointestinal side effects [38,39,40]. In
this context, further investigation remain to be performed.
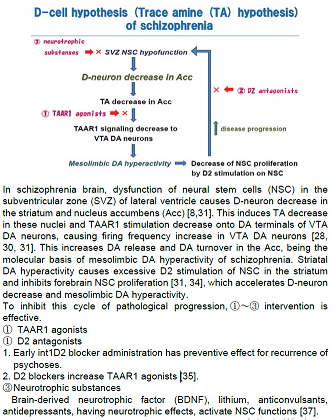
Figure 1: Scheme of D-cell hypothesis (trace amine (TA) hypothesis) of schizophrenia.
Some
Evidences Supporting D-Cell Hypothesis of Schizophrenia (Table 1)
Although it has not yet been detailed which type of TA in the human
central nervous system is related to psychiatric symptoms, nor has been
identified the endogenous ligands of human TAAR1, clinical and/or
pharmacological observations may enable us to determine the critical type of
TA. Further, the type of TA that is synthesized in human striatal D-neurons has
not yet been clarified. Early in 1974, Sabelli and Mosnaim proposed “Phenylethylamine
hypothesis of affective behavior” [41], indicating the involvement of TA in
animal behaviours. PEA, having the similar chemical structure of
methamphetamine, is the most probable TA which affects on psychiatric symptoms.
One of the initial clinical symptoms frequently observed in first episode schizophrenia
is the disturbance of sleep-wake-rhythm, that is, insomnia and daytime
hypersomnia. As PEA is the specific substrate for monoamine oxidase, type B
(MAOB), MAOB knockout mice contained elevated level of PEA in the striatum by 8-10
times of that of controls [42]. Clinically, MAOB inhibitor, selegiline
ameliorates daytime sleepiness of narcolepsy or other neuropsychiatric
diseases. This is explained by PEA increase due to inhibition of PEA
degradation. The D-neuron decrease in the striatum of schizophrenia [9] due to
NSC dysfunction,
causes striatal TA decrease. The authors post-mortem brain study has shown
increased DNA methylation rate of MAOB
gene in Acc of schizophrenia [43]. This may be the compensation for PEA
decrease caused by lack of D-neurons in Acc. From the aspect of food intake,
PEA is included in chocolate. High incidence of chocolate habit of Novel
Prizewinners, that is, eating chocolate more than twice a week, has been
reported [44]. PEA is supposed to be related to higher mental functions.
Whereas, too much chocolate intake of children is generally restricted, possibly
aimed at preventing D-neuron down regulation. Carlsson et al. reported that
administration of D2 antagonists such as chlorpromazine and haloperidol
increased TAAR1 ligands, including 3-methotytyramine and normetanephrine [36].
This indicates that the molecular basis of efficacy of D2 antagonists may be
effects also via TAAR1 stimulation by 3-methotytyramine and/ or
normetanephrine. Ventricular enlargement in brain imaging of patients with schizophrenia
[32,33] may be the similar phenomenon to D-neuron decrease in the striatum of
schizophrenia [9], both of which support NSC dysfunction hypothesis of
schizophrenia. Decreased level of plasma brain-derived neurotrophic
factor (BDNF) in schizophrenia [40] is also related to NSC dysfunction. Some
evidence supporting D-cell hypothesis of schizophrenia is summarized in Table
1.
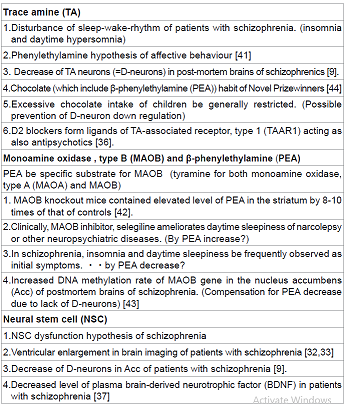
Table 1: Possible evidences supporting “D-cell hypothesis” (“Trace amine (TA) hypothesis”)
Prognoses
of Neuropsychiatric Illnesses
“D-cell
hypothesis”, which is proposed by a postmortem brain study of schizophrenia,
explains molecular mechanism of mesolimbic DA hyperactivity of schizophrenia, linking NSC dysfunction
hypothesis with DA hypothesis. Such D-cell-involved etiological dynamism in
schizophrenia may exist in wide spectrum of mental illnesses, and also in neurological
illnesses [45]. As shown in Figure 1, NSC functions affect not only on
D-neuron activity, but also clinical course and prognoses of neuropsychiatric
illnesses.
Conclusion
The D-neuron, i.e., the TA neuron, is a clue for
pathogenesis of neuropsychiatric illnesses. Exploration of endogenous TAAR1 ligands,
and NSC- and D-neuron-mediated signal transduction of normal and/or disease
state(s) is critical for future direction of neuropsychiatric
research.
Acknowledgement
The present study
was supported by Grant-in-Aid for Scientific Research from Japan Society for
the Promotion of Science (C-22591265, “How are trace amines involved in pathophysiology
of schizophrenia?”).
References
1.
Hökfelt T, Ljungdahl A, Fuxe K, Johansson O. Dopamine nerve terminals in the
rat limbic cortex: aspects of the dopamine hypothesis of schizophrenia (1974)
Science 184: 177-179.
2.
Toru M, Nishikawa T, Mataga N, Takashima M. Dopamine metabolism increases in
post-mortem schizophrenic basal ganglia (1982) J Neural Transm 54: 181-191.
3.
Watis L, Chen SH, Chua HC, Chong SA, Sim K. Glutamatergic abnormalities of the
thalamus in schizophrenia: a systematic review (2008) J Neural Transm (Vienna)
115: 493-511.
4.
Olbrich HM, Valerius G, Rüsch N, Buchert M, Thiel T, Hennig J, et al.
Frontolimbic glutamate alterations in first episode schizophrenia: evidence from
a magnetic resonance spectroscopy study (2008) World J Biol Psychiatry 9:
59-63.
5.
Christison GW, Casanova MF, Weinberger DR, Rawlings R, Kleinman JE. A
quantitative investigation of hippocampal pyramidal cell size, shape, and variability
of orientation in schizophrenia (1989) Arch Gen Psychiatry 46:1027-1032.
6.
McGlashan TH, Hoffman RE. Schizophrenia as a disorder of developmentally
reduced synaptic connectivity (2000) Arch Gen Psychiatry 57: 637-648.
7.
Duan X, Chang JH, Ge S, Faulkner RL, Kim JY, et al. Disrupted-In-Schizophrenia
1 regulates integration of newly generated neurons in the adult brain (2007)
Cell 130: 1146-1158.
8.
Reif A, Fritzen S, Finger M, Strobel A, Lauer M, et al. Neural stem cell proliferation
is decreased in schizophrenia, but not in depression (2006) Mol Psychiatry 11:
514-522.
9.
Ikemoto K, Nishimura A, Oda T, Nagatsu I, Nishi K. Number of striatal D-neurons
is reduced in autopsy brains of schizophrenics (2003) Leg Med (Tokyo) 5 Suppl
1: S221-224.
10.
Jaeger CB, Teitelman G, Joh TH, Albert VR, Park DH, et al. Some neurons of the
rat central nervous system contain aromatic-L-amino-acid decarboxylase but not
monoamines (1983) Science 219: 1233-1235.
11.
Boulton AA. Amines and theories in psychiatry (1974) Lancet 2: 52-53.
12.
Boulton AA, Juorio AV. The tyramines: are they involved in the psychoses? (1979)
Biol Psychiatry. 14: 413-419.
13.
Jaeger CB, Ruggiero DA, Albert VR, Park DH, Joh TH, et al. Aromatic L-amino
acid decarboxylase in the rat brain: immunocytochemical localization in neurons
of the brain stem (1984) Neuroscience 11: 691-713.
14.
Tashiro Y, Kaneko T, Sugimoto T, Nagatsu I, Kikuchi H, et al. Striatal neurons
with aromatic L-amino acid decarboxylase-like immunoreactivity in the rat
(1989) Neurosci Lett 100: 29-34.
15.
Mura A, Linder JC, Young SJ, Groves PM. Striatal cells containing aromatic L-amino
acid decarboxylase: an immunohistochemical comparison with other classes of
striatal neurons (2000) Neuroscience 98: 501-511.
16.
Ikemoto K, Kitahama K, Jouvet A, Arai R, Nishimura A, et al. Demonstration of
L-dopa decarboxylating neurons specific to human striatum (1997) Neurosci Lett
232: 111-114.
17.
Ikemoto K, Nagatsu I, Kitahama K, et al. A dopamine-synthesizing cell group
demonstrated in the human basal forebrain by dual labeling immunohistochemical
technique of tyrosine hydroxylase and aromatic L-amino acid decarboxylase
(1998) Neurosci Lett. 243: 129-132.
18.
Kitahama K, Ikemoto K, Jouvet A, Nagatsu I, Sakamoto N, et al. Aromatic L-amino
acid decarboxylase and tyrosine hydroxylase immunohistochemistry in the adult
human hypothalamus (1998) J Chem Neuroanat. 16: 43-55.
19.
Kitahama K, Ikemoto K, Jouvet A, Araneda S, Nagatsu I, et al. Aromatic L-amino
acid decarboxylase-immunoreactive structures in human midbrain, pons, and
medulla (2009) J Chem Neuroanat 38: 130-140.
20.
Ikemoto K. Significance of human striatal D-neurons: implications in neuropsychiatric
functions (2004) Prog Neuropsychopharmacol Biol Psychiatry 28: 429-434.
21.
Bunzow JR, Sonders MS, Arttamangkul S, Harrison LM, Zhang G, et al. Amphetamine,
3,4-methylenedioxymethamphetamine, lysergic acid diethylamide, and metabolites
of the catecholamine neurotransmitters areagonists of a rat trace amine
receptor (2001) Mol Pharmacol 60: 1181-1188.
22.
Borowsky B, Adham N, Jones KA, Raddatz R, Artymyshyn R, et al. Trace amines:
identification of a family of mammalian G protein-coupled receptors (2001) Proc
Natl Acad Sci U S A 98: 8966-8971.
23.
Miller GM. The emerging role of trace amine-associated receptor 1 in the functional
regulation of monoamine transporters and dopaminergic activity (2011) J
Neurochem 116: 164-176.
24.
Xie Z1, Miller GM. Trace amine-associated receptor 1 as a monoaminergic modulator
in brain (2009) Biochem Pharmacol 78: 1095-1104.
25.
Lindemann L, Meyer CA, Jeanneau K, Bradaia A, Ozmen L, et al. Trace amine-associated
receptor 1 modulates dopaminergic activity (2008) J Pharmacol Exp Ther 324:
948-956.
26.
Zucchi R, Chiellini G, Scanlan TS, Grandy DK. Trace amine-associated receptors
and their ligands (2006) Br J Pharmacol 149: 967-978.
27.
Bradaia A, Trube G, Stalder H, et al. The selective antagonist EPPTB reveals
TAAR1-mediated regulatory mechanisms in dopaminergic neurons of the mesolimbic
system (2009) Proc Natl Acad Sci USA. 106: 20081-20086.
28.
Revel FG, Moreau JL, Hoener MC, et al. A new perspective for schizophrenia: TAAR1
agonists reveal antipsychotic- and antidepressant-like activity,improve
cognition and control body weight (2013) Mol Psychiatry. 18:543-556.
29.
Panas HN, Lynch LJ, Vallender EJ, et al. Normal thermoregulatory responses to 3
iodothyronamine, trace amines and amphetamine-like psychostimulants in trace
amine associated receptor 1 knockout mice (2010) J Neurosci Res. 88: 1962-1969.
30.
Wolinsky TD, Swanson CJ, Smith KE, et al. The Trace Amine 1 receptor knockout
mouse: an animal model with relevance to schizophrenia (2007) Genes Brain
Behav. 6: 628-639.
31.
Sanai N, Tramontin AD, Quiñones-Hinojosa A, Barbaro NM, Gupta N, et al. Unique
astrocyte ribbon in adult human brain contains neural stem cells but lacks
chain migration (2004) Nature 427: 740-744.
32.
Degreef G, Ashtari M, Bogerts B, et al. Volumes of ventricular system subdivisions
measured from magnetic resonance images in first-episode schizophrenic patients
(1992) Arch Gen Psychiatry. 49: 531-537.
33.
Horga G, Bernacer J, Dusi N, et al. Correlations between ventricular enlargement
and gray and white matter volumes of cortex, thalamus, striatum, and internal
capsule in schizophrenia (2011) Eur Arch Psychiatry Clin Neurosci. 261:
467-476.
34.
Kippin TE, Kapur S, van der Kooy D. Dopamine specifically inhibits forebrain
neural stem cell proliferation, suggesting a novel effect of antipsychotic drugs
(2005) J Neurosci. 25: 5815-5823.
35.
Carlsson A, Lindqvist M. Effect of Chlorpromazine or Haloperidol on formation
of 3 methoxytyramine and normetanephrine in mouse brain (1963) Acta Pharmacol
Toxicol (Copenh) 20: 140-144.
36.
Penttilä M, Jääskeläinen E, Hirvonen N, Isohanni M, Miettunen J. Duration of
untreated psychosis as predictor of long-term outcome in schizophrenia: systematic
review and meta-analysis (2014) Br J Psychiatry 205: 88-94.
37.
Fernandes BS, Steiner J, Berk M, et al. Peripheral brain-derived neurotrophic factor
in schizophrenia and the role of antipsychotics: meta-analysis and implications
(2015) Mol Psychiatry 20: 1108-1119.
38.
Piazza J, Hoare T, Molinaro L, Terpstra K, Bhandari J, et al. Haloperidolloaded
intranasally administered lectin functionalized poly(ethylene glycol)- block-poly(D,L)-lactic-co-glycolic
acid (PEG-PLGA) nanoparticles for the treatment of schizophrenia (2014) Eur J
Pharm Biopharm 87: 30-39.
39.
Wen Z, Yan Z, Hu K, et al. Odorranalectin-conjugated nanoparticles: preparation,
brain delivery and pharmacodynamic study on Parkinsons disease following
intranasal administration (2011) J Control Release. 151:131-138.
40.
Ikemoto K, Nishi K, Nishimura A. Lectin-positive spherical deposit (SPD) in the
molecular layer of hippocampal dentate gyrus of dementia, Downs syndrome,
schizophrenia (2014) J Alzheimers Dis Parkinsonism. 4: 1000169. 41. Sabelli HC,
Mosnaim AD. Phenylethylamine hypothesis of affective behavior (1974) Am J
Psychiatry 131: 695-699.
42.
Grimsby J, Toth M, Chen K, Kumazawa T, Klaidman L, et al. Increased stress
response and beta-phenylethylamine in MAOB-deficient mice (1997) Nat Genet 17:
206-210.
43.
Yang QH, Ikemoto K, Nishino S, et al. DNA methylation of the monoamine oxidases
A and B genes in postmortem brains of subjects with schizophrenia (2012)
OJPsych. 2: 374-383.
44.
Golomb BA, Brenner S, Chalfie M, Glashow SL, Glauber RJ, et al. Chocolate habits
of Nobel prizewinners (2013) Nature 499: 409.
45.
Bachmann RF, Schloesser RJ, Gould TD, Manji HK. Mood stabilizers target cellular
plasticity and resilience cascades: implications for the development of novel
therapeutics (2005) Mol Neurobiol. 32: 173-202.
*Corresponding author
Keiko Ikemoto, Department of Psychiatry, Iwaki Kyoritsu General Hospital, Iwaki 973-8555, Japan, Tel:+81-246-26-3151 Fax:+81-246-27-2148 E-mail: ikemoto@iwaki-kyoritsu.iwaki.fukushima.jp
Citation
Ikemoto K (2015) D-Cell Hypothesis (Trace Amine Hypothesis) of Schizophrenia, and importance of Trace Amine-Associated Receptor, Type 1 (TAAR1). PVPE 102: 1-5