

Review Article :
Y Anu Shanu, Antonio Lauto, Simon J Myers Coactosin is one of the numerous actin-binding
proteins which regulate the actin cytoskeleton. Coactosin binds F-actin, and
also interacts with 5-lipoxygenase, which is the first committed enzyme in
leukotriene biosynthesis. Coactosin and human coactosin like protein 1 (COTL1)
have the potential to play a role in the degradation or impairment of neuronal
cells and their functioning. Its homology to other proteins that affect
neuronal cells also contributes to this notion. The objective of this review is
to explore its structural novelty, regulation and its significance in
neurodegenerative diseases. Neurodegenerative
disorders include some of the most common and least understood diseases
characterised by impaired sensory motor and/or mental functioning. Many such as
Alzheimers disease currently have no cure and subsequently have become the
focus of significant research in searching for an understanding of the
molecular pathophysiology of the disease and looking for methods of prevention
treatment and cure. Neurodegenerative
disorders are categorised by the loss of structure and/or function of neurons.
Neuronal cell degeneration and death as well as plaques and atypical protein
assemblies are the most common causes of such loss of structure and function
(Heemels 2006). Neurons are made up of two morphologically defined regions: the
dendrites and the axons. Microtubules actin filaments and neurofilaments make
up the neuronal cytoskeleton and are responsible for several critical roles
including the formation of axons intracellular transport of cargoes and
maintenance of its structure (Kevenaar &Hoogenraad 2015). Coactosin is a 17
kDa actin binding protein originally isolated from Dictyostelium discoidum but it has since been isolated from other
organisms including humans fruit flies and mice (Poukkula et al. 2011). Both coactosin and human coactosin like
protein 1 (COTL1) can bind to 5-lipoxygenase (5-LO) and filamentous actin
(F-actin) (Lackie 2007) and has the potential to play a role in the degradation
or impairment of neuronal cells and their functioning. Its homology to other
proteins that affect neuronal cells also contributes to this notion. Coactosin shows
structural homology to actin-depolymerising factor (ADF-H) domain. The ADF
homology domain a highly conserved protein motif promotes cytoskeletal dynamics
by facilitating processes such as endocytosis cell
migration morphogenesis and cytokinesis (Poukkula et al. 2011) owing to its binding affinity towards filamentous
actin (F-actin) and not globular actin (G-actin). ADF domain consists of five
internal β-sheets of which β1- β4 are antiparallel and β4 is parallel to β5.
The sheets are surrounded by four α-helices with α1-α3 parallel to the β-sheets
and α4 packed perpendicular to the β3 and β4 sheets (Hellman et al. 2004). Despite the
similar structure the amino acid sequence of COTL1 shows low homology to
Coactosin with 33.3% sequence identity and 75% similarity (Liu et al. 2004).
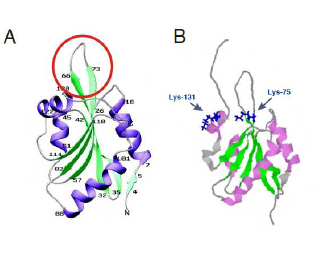
COTL1 is entirely composed of a single ADF domain and consists of five
β-stranded sheets with two helices each on either side (Fig. 1A). The loop between β3-β4 is flexible while the
other regions are held rigid (Rakonjac 2009). Lys-75 and Lys-131 which
corresponds to binding sites for F-actin and 5-LO respectively (Fig. 1B) lies
close to each other suggesting overlapping binding sites which has been
suggested as a structural mechanism to prevent simultaneous binding (Liu et al
2004) which is also supported by the fact that a 5-LO-COTL1-F-Actin ternary
complex has not yet been reported (Esser et al. 2010). Figure 1: Structure
of human COTL1. COTL1 plays
several functional roles in normal physiology however it is primarily
identified as an actin binding protein due to its role in the promotion of
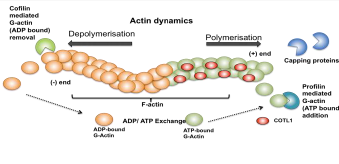
actin polymerisation. Actin treadmilling is a dynamic process where ATP bound
globular monomer actin (G-Actin) is added on to the + end of the growing
filament by ADF-H protein Profilin promoting actin polymerisation (Fig. 2)
where as depolymerisation occurs when Cofilin another ADF-H protein removes ADP
bound G-Actin from the - end of the filament (Hou et al. 2013). Capping
proteins are employed at the + end to regulate the rate of polymerisation and
COTL1 counteracts the activities of the capping proteins cap32/34 (Rohrig et
al. 1995). Therefore by interfering with these proteins COTL1 indirectly
promotes actin polymerisation (Hou et al. 2013 Liepinsh et al. 2004 Rohrig et
al. 1995). The binding follows a 1:2 stoichiometry between COTL1 and actin and
is a Ca2+ independent process (Rakonjac 2009). ATP-G-actin is
added on to the + end by Profilin promoting actin polymerisation and
ADP-G-actin is removed from the - end by Cofilin resulting in depolymerisation.
COTL1 binds to F-actin at a 1:2 ratio preventing binding of capping proteins
thereby promoting actin polymerisation.
This dynamic
polymerisation-depolymerisation process enables F-actin to perform its
functions involving cell migration and morphogenesis. Growth cones are
structures that lead axons to synaptic targets by virtue of this process (Hou
et al. 2013). Actin polymerisation drives the protrusion of lamellipodia and
filopodia in the growth cones of axons (Gomez & Letourneau 2013). Therefore
COTL1 which functions in actin polymerisation regulation plays a critical role
in neurite extension and neural crest migration (Hou et al. 2013). COTL1 was
also found to be significantly involved in T-cell migration following CD28
stimulation (Kim et al 2014). Suppression of COTL1 prevented lamellipodial
protrusion at the T cell – B cell contact site owing to impaired T cell
spreading in response to TCR ligation (Kim et al 2014). Figure 2: Schematic
diagram showing the dynamic nature of F-actin. Role of COTL1 in Leukotriene
synthesis Both coactosin
and COTL1 can bind to 5-LO to promote the formation of leukotrienes (LTs) lipid
mediators that elicits inflammatory responses in allergic reactions asthma and
atherosclerosis (Funk 2005). Since inflammatory reactions are part of cell
defence mechanisms as well as chronic pathologies it is imperative that
regulation of 5-LO is a key step in maintaining the balance. Any insult
resulting in an increase in intracellular Ca2+ leads to the
association of COTL1 to 5-LO in a 1:1 molar stoichiometry prompting the
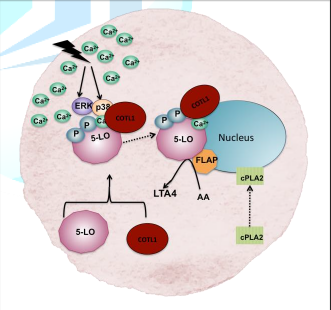
translocation of the 5-LO-COTL1 complex to the perinuclear area (Fig. 3) where
it docks to nuclear membrane phosphatidylcholine (PC) with the help of 5-LO
activating protein (FLAP) (Esser et al. 2010 Liepinsh et al. 2004). A cellular
stimuli leading to high intracellular Ca2+ or activation by MAP
kinases ERK or p38 is thought to induce association of COTL1 with 5-LO. FLAP
assists the translocation of the 5-LO- COTL1 complex to the nuclear membrane
where it converts AA to LTA4 (Adapted from Basavarajappa et al. 2014 and
Rakonjac 2009). The enzyme cytosolic phospholipase A2
(cPLA2) which is also translocated from cytosol to perinuclear area
releases arachidonic acid (AA) from PC which is accessed by the 5-LO complex
and converted into LTA4 (Basavarajappa et al. 2014). 5-LO catalyses
two initial steps in LT formation the oxygenation of arachidonic acid to
5(S)-hydroperoxy-681114-eicosatetraenoic acid (5-HPETE) and the dehydration
into epoxide. Thus although 5-LO appears to be the initiator of the LT pathway
complete cellular 5-LO activity in presence of Ca2+ including the
nuclear translocation was displayed only in presence of COTL1 and was absent in
a COTL1 knockdown model (Basavarajappa et al. 2014). LTA4 undergoes
further enzymatic modifications to form either LTB4 or cistenyl
leukotrienes LTC4 LTD4 or LTE4 (Rakonjac
2009). Both LTB4 andcistenyl leukotrienes have been
found to induce monocyte chemo attractant protein 1 (MCP-1) a chemokine heavily
involved in neuroinflammatory responses (Huang et al 2004 Ichiyama et al 2005). Taken together these data
invariably suggest the significance of 5-LO-COTL1 association in
neuroinflammation and potentially neurodegeneration. In addition to increased
intracellular Ca2+ other stress stimuli causing an increase in p38
and ERK MAP kinase activation can subsequently phosphorylate 5-LO leading to
its activation and ensuing LTA4 production (Rådmark and Samuelsson 2009). There is a lack
of research conducted on the direct interaction of coactosin and COTL1 on
organelles outside of the cytoskeleton. However there is an understanding on
how cofilin of which coactosin shares homology interacts with mitochondria. Cofilin plays a
role in the regulation of mitochondrial action in apoptosis (Li et al 2013).
Apoptotic cell death is characterised by a distinct change in cellular
architecture demonstrated by blebbing of plasma
membrane externalization of phosphatidylserine nuclear condensation and finally
DNA fragmentation and release of cellular contents into circulation. Apoptosis
occurs in three stages. The first is initiation in which a stress occurs that
causes activation of one of various pathways to trigger a death signal that
once sensed by the appropriate receptor leads to the second stage. The second
stage is the commitment stage. Once a cell reaches this point the cell cannot
reverse the process and will proceed to death. During this stage in apoptosis
apoptotic proteins interact with the mitochondria to permeabilise it to allow
effectors such as cytochrome c to leave. Dephosphorylated Cofilin (Ser3)
translocates from the cytosol to the mitochondria and opens pores in the
mitochondria to allow cytochrome c out thereby playing a role in the commitment
stage of apoptotic cell death (Li et al 2013). Cytochrome c then forms a
complex with activating factor-1 to cleave caspase-9 which activates Caspase-3.
This step is considered part of the third stage of cell death the execution
stage as it cleaves proteins that are essential to normal cell functioning.
This leads to apoptosis resulting in cell death. It has also been shown that
knockdown of cofilin leads to a reduction in the release of cytochrome c and
hence apoptosis and cell death (Li et al. 2013 Li et al. 2015 Taha Mullen &
Obeid 2007). Sphingolipids are
a class of lipids that contain a long chain sphingoid base (for example
sphingosine). They can be found in mammalian plasma membrane and have been
shown to play a role in a variety of cell signaling events including cell growth
cell death cell differentiation and stress responses (Shayman 2000 Taha Mullen
& Obeid 2007). Ceramides are a
class of sphingolipids with a fatty acid linked to the amide group of the
long-chain base. They serve as an intermediate for more complex sphingolipids
such as phosphosphingolipids and glucosphingolipids. It has been proposed that
ceramides play a role in signaling for programmed cell death based on two
primary observations. The first is that agonists that induce apoptosis such as
CD95 and chemotherapeutic drugs raise cellular levels of ceramide. The second
is the artificial addition of cell permanent ceramides leads to an apoptotic
response (de Chaves 2006). There is evidence
indicating the possibility that sphingolipids may play a role in
neurodegenerative disorders. An increased level of ceramides has indeed been
detected in the brain of Alzheimers patients in early stages along with a
reduced level of sulfatides in their brain grey and white matter and
cerebrospinal fluid (Han et al 2002). The elevated ceramide levels most likely
arose from the breakdown of the sulfatides and can therefore be a potential
biomarker for Alzheimers (de Chaves 2006).
Due to the role of sphingolipids in the
signalling of programmed cell death as well as its potential involvement in
neurodegenerative disorders it is possible that COTL1 may interact with this
biomolecule. An increase in COTL1 in endoplasmic reticulum fraction was in fact
reported in lymphocytes isolated from Hereditary Sensory Neuropathy Type
1(HSN1) patients expressing V144D mutation in their serine palmitoyltransferase (SPT) long chain subunit 1 (SPTLC1)
(Stimpson et al 2016a). This increase in COTL1 was also
further established in C133W C133Y and V144D mutants in a neuronal cell model
for HSN1 (Stimpson et al 2016). SPT is a key enzyme involved in sphingolipid
synthesis. The disease is characterised by degeneration of the dorsal root
ganglion and presents with clinical onset from the second or third decades of
the patient life (Stimpson et al 2016b). Although the
actual mechanism of COTL1 regulation is yet to be elucidated its upregulation
could be instigated by the increased oxidative stress upon the cellular
cytoskeletal system which can cause actin remodelling and potential axonal
retraction in the neuron (Stimpson et al 2016a). Increased COTL1
might also be triggering inflammatory pathways owing to its 5-LO binding
property and subsequent LTA4 synthesis as described earlier. Whether COTL1 has
any direct effects on sphingolipid metabolism or vice versa needs to be further
investigated. Currently there
are very few other published reports on sphingolipid and COTL1 interaction.
There is however one report that includes the interaction of a ceramide with
coflilin-1 and F-actin in the stimulation of mouse embryonic stem cell
migration (Park et al. 2013). The report indicates that the ceramide promoted
the interaction between cofilin and F-actin to enhance mouse embryonic stem
cell migration. This serves to further implicate a potential interaction
between sphingolipids and COTL1. In Alzheimers and
other neurodegenerative disorders inflammatory processes (such as the ones that
COTL1 help to regulate) play a crucial role. 5-LO plays an important role in
inflammatory responses which are triggered by the presence of plaques such as
in Alzheimers patients. This results in activation of microglia and astrocytes
leading to neuron cell degeneration and death. This can however cause worsening
of the disease rather than healing it. Furthermore it has been shown that
downregulation of 5-LO improves the memory and synaptic functioning in animal
Alzheimers models. Also there has been some epidemiological evidence that may
suggest that anti-inflammatory treatment can have a positive effect in
preventing or minimising the effect of Alzheimers (Czapski et al. 2016). Figure 3: Schematic
diagram showing 5-LO activation, translocation and LTA4 synthesis in
the cell. Coactosin and
COTL1 have ADF/cofilin homology. This family of proteins is essential in the
formation of neurites via the organisation of actin filaments (Sainath &
Gallo 2015). For example cofilin increases actin depolymerisation and
facilitates actin filament turnover as a way to regulate actin dynamics. Rods form spontaneously in neurons that
overexpress ADF/cofilin disrupting the microtubules. This does not cause cell
death but does cause degeneration leading to a loss of synapses resulting in
neurodegeneration (Minamide et al. 2000). Cofilin
aggregates and actin bundles have been observed in Alzheimers brains. In low
ATP environment (such as one caused by mitochondrial dysfunction or oxidative
stress) there tends to be a higher ADP-actin and dephosphorylated cofilin
concentration. Under these conditions the cofilin-actin complex readily forms
into rods (neuropil thread structures). Rods that form as a result to this low
ATP condition usually disappear shortly after however in neurites with
irreversible mitochondrial damage these rods become more permanent and can lead
to loss of synaptic connections without loss of cells as mentioned above. This
could be an explanation for the similar conditions that have been reported to
occur in the early stages of neurodegenerative conditions (Maloney &
Bamburg 2007 Whiteman et al. 2009). Smith-Magenis
syndrome (SMS) is caused by deletion of the short arm of chromosome 17. Its
symptoms include neuro-behavioural abnormalities and congenital heart defects.
The COTL1 gene is mapped to SMS common deletion region. This may indicate COTL1s
involvement in the disease. The region also overlaps with a region associated
with primitive neuro-ectodermal tumours. This suggests that COTL1 plays a role
in DNA rearrangements of somatic cells (Nakatsura et al. 2002). Upregulated COTL1 expression has been
identified in small cell lung cancer tissue thus suggesting it may be a
biomarker or therapeutic target in these cancer patients (Jeong et al. 2010). Neurodegenerative
disorders are those that affect the normal functioning of the brain and its
components. This occurs as a result of degradation of neurons including the
formation of plaques and/or atypical protein assemblies. Despite being very
common very few of these disorders have a cure. As a result there is a large
amount of research dedicated to understanding and finding cures or even early
detection markers for these various diseases. One such example is the research
conducted on the effect of COTL1 on neurodegeneration. COTL1 being an
actin binding protein plays a role in the maintenance of neuron structure
through its interaction with the cytoskeleton and hence can potentially be
involved in the loss of proper neuron structure leading to neurodegeneration.
It is also involved in leukotriene synthesis for inflammatory responses which
can be triggered by plaques in patients suffering from these neurodegenerative
disorders and potentially worsens the condition. COTL1 shares homology with the
ADF/cofilin family of proteins that play a role in mitochondrial apoptosis.
This apoptotic effect can lead to unnecessary cell death in a low ATP
environment and hence cause a large unwarranted loss of neurons and/or the
formation of neuropil thread structures which can also lead to
neurodegeneration. Furthermore sphingolipids also play a role in apoptosis and
other important cell signalling events. The interaction between them and COTL1
may also be important in the triggering or control of neurodegeneration. Currently there
exists little research on the direct effect of COTL1 on neurodegeneration but
what little there is does show promise in there being a link between the two.
Subsequently more research needs to be conducted to further understand the
connection if any between COTL1 and neurodegeneration. COTL1, Coactosin, 5 Lipoxygenase, Actin polymerisation, Hereditary Sensory Neuropathy, Neurodegeneration, Cytoskeleton
The Role of Human Coactosin-Like Protein in Neurodegenerative Disorders
Abstract
Full-Text
Structure of COTL1
COTL1 and Actin polymerisation
Interaction of COTL1 with
Organelles
Interaction of COTL1 with Sphingolipids
The Role of COTL1 in
Neurodegeneration
Conclusion
References
Keywords
