Poly
lactic-co-glycolic acid, referred to as PLGA, is one of the most
successfully used biodegradable polymers used in controlled drug delivery systems [1-4]. Over the
past 50 years, the development of biodegradable polymers has represented a
revolution in medicine and has led to significant biotechnological advancements
for drug delivery, biomaterials, tissue
engineering, and medical device
development. The
development of these biodegradable polymers has been made possible through a
unique collaboration between chemists, engineers, biologists, and physicians.
One of the major driving forces for the development of polymeric drug delivery
platforms has been the necessity of improving cancer therapeutics. Currently,
anti-cancer drugs have short half-lives, nonspecific drug distribution
throughout the body, and acute toxicity to non-malignant cells [5].
For the treatment of
prostate cancer, controlled-release nanodrugs delivery platforms have substantial
advantages, compared to conventional treatments, because they can overcome pharmacological limitations
such as drug resistance. Polymeric NP drug delivery systems possess the
capacity for localized and sustained drug delivery, as well as the ability to
improve the therapeutic index of various drugs. The numerous therapeutic
advantages of polymeric drug delivery platforms can be attributed to their
versatile nature and ability to control drug release [6-8].
A common endeavor in
nanomedicine has been the encapsulation of NPs with PLGA polymer [9]. Polyesters, such as PLGA, have been
approved by the FDA and EMA, and are
generally well tolerated within the body [10,11]. Due to this, polyesters are
the most commonly investigated class of biodegradable drug delivery systems [12].
Much of the current interest in NPs as drug
delivery vehicles has arisen from the potential of NPs to increase pharmacokinetic activities and improve the safety
profiles of the cargo (therapeutic drugs) in which they encapsulate. Numerous NP delivery system formulations
are under clinical evaluation, while several have already been translated into
clincical application and are available on the market [13,14]. Many of these nano formulations are being developed for oncological use because NPs can “passively” accumulate
within tumors through a phenomenon known as Enhanced Permeability And Retention
(EPR) effect [15]. NPs accumulate through
EPR by exploiting defects in the neo vasculature endothelial junctions and
impaired lymphatic drainage. Functionalizing
the surface of NPs with targeting ligands can further enhance cellular uptake
and tumor site retention through a concept known as “active” targeting [16]. NPs
are promising new drug carrier systems due to their exceptional
biocompatibility as well as their ability to control and sustain the release of
drugs [17,18].
The potential to solubilize poorly soluble
therapeutic substances, reduce drug toxicity, prolong drug circulation time,
control drug release kinetics, improve drug targeting, and enhance therapeutic efficacy
through monitored drug delivery, has encouraged the continual expansion of this
type of research [19-27]. NPs which possess the correct size, shape, and cell
surface properties can systemically circulate for prolonged periods of time,
“passively” target cancerous tumors through accumulation using the EPR effect,
and locally release the drug to malignant cells [28-34]. NP drug delivery systems
have promising potential for reducing the development of Multidrug Resistance
(MDR) during prostate cancer treatment through controlled chemotherapeutic drug
release at the site of the malignancy [25-27].
The intervention of nanoparticle
drug delivery is needed because Prostate Cancer (PC) is the most commonly
diagnosed male malignancy in the western world [35]. The development of drug
resistance and progression to
metastasis are common clinical implications of those who are actively managing
PC. For PC to metastasize to distant sites throughout the body, PC cells must
first migrate and invade neighboring tissue(s). Malignant cells, including PC
cells, can acquire a migratory and invasive phenotype by various means
including single
cell and collective cell
migration. Additionally,
a motile, mesenchyme-like phenotype is often required for PC cell migration. To
acquire this phenotype, polarity and epithelial characteristics (example,
expression of E-cadherin homotypic adhesion receptor) frequently have to be
lost as well, mesenchyme phenotypic characteristics (for example, cytoskeletal
rearrangements, enhanced expression of proteolytic
enzymes and other repertory of
integrin’s) have to be developed. The entire process is known as the
Epithelial-To-Mesenchyme Transition (EMT).
One of the hallmarks of
cancer is cellular invasion. Cellular invasion is defined by the movement of
cells through a three-dimensional matrix, resulting in cellular environment
remodeling. The essential components of cellular invasion are cellular
adhesion, proteolysis of the ECM, and malignant cell migration. In-vitro
studies on the migratory and invasive abilities of cells are useful tools for
assessing the aggressiveness of solid tumors, including those of the prostate.
The Trans well migration assay (a common in-vitro
technique used to investigate the migratory behavior of PC cells) was
introduced in this study as an alternative method for quantifying the amount of
SC-514 released from the SC-514-PLGA NPs.
The NP encapsulation method
utilized can influence the amount of drug released and the effectiveness of
drug quantification method(s). The methodology and material used for
encapsulating poorly soluble, fragile, or toxic compounds is vital for drug
delivery. By bettering the efficacy of drug encapsulation in drug carrier
particles, stronger therapeutic effect(s) and minimalized negative side
effect(s) can be achieved [36]. As a result, examining the potentiality of new
encapsulation materials and understanding the various drug-carrier interactions (the interaction between
the drug and the encapsulating material) permits the development of new
methods. Drug-carrier interactions have the ability to significantly increase
the entrapment of the drug and, thus, are of further importance when
considering drug design [36].
The construction of
drug-controlled delivery
systems for the treatment of
various diseases, including cancer, is of significant research interest due to
their facilitation of high therapeutic efficacy, avoidance of repeated drug
administrations, and betterment of patient compliance [37,38]. Many
drug-release systems are also sensitive to external stimuli such as
temperature, pH, magnetism, or electric fields [39-41]. These Stimuli-responsive drug carriers can release their encapsulated
drug in a controlled manner compared to that of conventional drug delivery
systems. Drug delivery systems
encapsulated with polymers, including PLGA, have demonstrated the capacity for
this controlled release performance [42-44]. The
solubility of a drug is generally intrinsically related to the drug’s particle
size - as a particle becomes smaller, the ratio of surface area to volume
increases. The larger surface area of small particles allows for greater
interaction with the solvent, resulting in increased solubility [45].
Nano therapeutics can be
exploited for the delivery of poorly soluble compounds, such as SC-514, through
intravenous drug administration. SC-514 is a relatively new, small molecule
drug that has potential therapeutic use for the treatment of prostate
cancer [46]. However, due to the
poor solubility of the compound, SC-514 is classified as a class IV or class II
drug, according to Bio Pharmaceutics Classification Systems (BCS)
classification [47]. NPs encapsulated with PLGA are potentially compelling
delivery systems for optimizing the conditions for SC-514 drug delivery,
solubility, and controlled release into tissues and cells, by protecting the
drug from oxygen and acids [48]. In this study, PLGA encapsulated NPs were
utilized to improve the bioavailability of the poorly water-soluble, SC-514.
NPs that have accumulated at
the target site require changes to their drug release rate in order to improve
their efficacy [49-51]. When formulating a NP carrier for a drug it is
important to consider optimizing drug loading, and quantifying the amount of
drug that remain associated with the carrier over various points in time [52].
NP-drug formulation performance is partially dependent on the efficiency of
drug loading, which is often determined by the Encapsulation
Efficiency (EE), EE is the percentage
or fraction of drug that is associated with the NP carrier after particle
manufacture and during drug release. The time course of NP drug release is an
additional principal factor because it establishes the amount of free drug
available over time.
The availability of free
drug is essential for therapeutic effect and, occasionally, for modifying the
drug’s toxicity profile [53,54].The in-vitro
drug release profiles (drug loading and drug release efficiency), measured in
bio-relevant medium, can provide substantial predictive evidence for in-vivo behavior of the encapsulated
drug as well as the mechanism(s) of drug
release [52,55]. Insight into the
mechanism(s) of drug release can further be utilized during formulation
parameter optimization to achieve the desired release rate properties, such as
NP surface area. Thus, investigating the in-vitro
drug release kinetics of NP-drug formulations is essential for proper
nanoparticle design and in-vitro−in-vivo correlations.
The drug release mechanisms
of NP carriers can be chosen based on the biological differences between the
tumor microenvironment and healthy tissue; These differences include lower pH,
lower oxygen levels, increased matrix metalloproteinase enzymatic activity, and
variance in NF-κβ activation [56,57]. In the tumor microenvironment, NF-κβ is
the primary transcription factor involved in immune system function regulation
and plays a critical role in cancer development and progression [57].
Additionally, NF-κβ regulates various
biological activities including cell proliferation and differentiation.
Activation of NF-κβ is correlated with proliferation of hematopoietic stem
cells and resistance to apoptosis.
These contrasting activities seemingly occur through a balance of the
transcription factor’s biological and biochemical functions [58,59].
Furthermore, NF- κβ has a well-defined role in oxidative stress as it increases
Nitric Oxide (NO) through Inducible Nitric Oxide Synthase (iNOS) activation.
Although acute NO production can trigger apoptosis, and the process of iNOS
activation is often regarded as part of NF- κβ’s pro-apoptotic function, the
continuous production of NO, due to constant activation of NF- κβ, may
potentially inhibit apoptosis [59-62].
Up regulation of
anti-apoptotic NF-κB target genes have been reported in various types of
malignant tumors. Among these genes are, Inhibitors of Apoptosis (IAPs), FLICE-like
inhibitory protein (FLIP), and members of the
B cell-lymphoma 2 (Bcl-2) family that inhibit apoptosis [63-65]. NF-κB
activation has also been associated with the up regulation of enhancers
involved in cell proliferation (i.e., Cyclic D1 and Cellular Myelocytomatosis
(c-myc)) and cell adhesion molecules, as well as angiogenic factors that
enhance malignant cell engraftment (i.e., Intercellular Adhesion Molecule 1
(ICAM-1) and Vascular Endothelial Growth Factor (VEGF)) [63-68].
Furthermore, NF-κB
activation regulates the expression of heme oxygenase-1 (HO-1), a catabolic
enzyme that acts on the free heme group [69]. Enhanced free heme catabolism
(increased HO-1 activity) has a protective role against apoptosis because free
heme is known to cause damage to the lipid bilayer of the cellular membrane [70].
In cancers, the up regulation of HO-1 has been shown to aid in evading
apoptosis induced by Tumor Necrosis Factor-α (TNF- α), as well as apoptosis
induced by chemotactic drugs [64]. Due to the implications of NF-κB in cancer
cell survival and progression,
this study investigated the potential impact of NF-κB signaling pathway
activation on PLGA-NP drug release, within the microenvironment of PCa cells.
To accomplish this, NF-κB antibodies (conjugant) were conjugated to the PLGA-NP
carrier systems.
Another factor utilized to
investigate the drug release of SC-514 in this study was fat accumulation
around PLGA-NP carrier systems. This was done because obesity is associated
with numerous chronic medical conditions and diseases, including prostate
cancer. In almost every country where detailed data is available, obesity has become more
pervasive [71]. Evidence has suggested that the prevalence of obesity has been
increasing for over one hundred years, however, in the United States, there
appears to be an accelerated rate of increase beginning around the 1980s [72-76].
Obesity has become an
epidemic as one in six American adults were considered to be obese 20 years
ago; yet one in three American adults are considered to be obese today [77-82].
The past twenty years of increased obesity prevalence has occurred throughout
every age, race, sex, and socioeconomic group, and is correlated to a decrease
in physical activity and an increase in poor dietary consumption [83-84]. Further,
fat deposition has been suggested to influence bioavailability and effect drug
release in the tumor microenvironment [85-86]. Due to this, the potential
impact of fat accumulation on the drug release profile of SC-514 from
SC-514-PLGA-Fat NPs was investigated in this study.
Surface modification of
nanoparticles is a key requisite for extending circulation half-life and
promoting localization. For example, nanoparticles coated with a highly
cationic polymer have been used to enhance cellular uptake or open
intercellular tight junctions [87,88]. Foliate receptors over-expressed on the
surface of malignant human
cells were targeted by grafting
foliate on the surface of nanoparticles [19]. Studies revealed that the
nanoparticles attained a 10-fold higher affinity for the surface foliate
binding protein than free foliate [89]. Researchers reasoned that the multivalent
form of foliate on the nanoparticle surface interacted strongly with foliate
receptors, which are often present in clusters on the surface of cancer cells,
like the clustering of ICAM-1 during T-cell
adhesion. Finally, research efforts
are ongoing to improve nanoparticle performance in-vivo by extending nanoparticle circulation and limiting
interaction with blood constituents [90] and in-vitro. However, it is not well understood how the kinetics of
such a drug delivery system will proceed [17] especially with SC-514 loaded
PLGA nanoparticles and conjugation of SC-514 loaded PLGA nanoparticles with
other molecules such as NF-KBAb and Fat. This conjugation may alter the
Encapsulation Efficiency (EE) and the drug release profile.
The measurement of both EE
and in-vitro drug release from
colloidal particles typically requires methods for the rapid physical
separation of particles from their surrounding dispersion medium to enable
real-time determination of the proportion of free drug. For large
particles this may be achieved by a
simple filtration approach. However, separation can be challenging for
nanoparticles due to their small size [91]. Most methods for the measurement of
encapsulation and in-vitro release
separate the particles from the medium in which they were dispersed and rely on
the quantification of the ‘free’ fraction of drug to indirectly measure the
nanoparticle-bound fraction. Numerous methods for the separation of free and
nanoparticle-associated drug are dialysis-based methods, ultracentrifugation,
centrifugal ultrafiltration and pressure ultrafiltration [91-96].
After SC-514 was released
from SC-514-PLGA nanoparticles, Liquid Chromatography-Mass Spectrometry (LC–MS)
was utilized as the standard method to quantify the SC-514 drug released from
SC-514-PLGA nanoparticles. Liquid chromatography-tandem mass spectrometry
(LC-MS/MS) has seen enormous growth in routine toxicology laboratories.
LC-MS/MS offers significant advantages over other traditional testing, such as
immunoassay and gas chromatography-mass spectrometry methodologies. Major
strengths of LC-MS/MS include improvement in specificity, flexibility, and
sample output when compared with other technologies [97]. The LC-MS/MS steps
usually involve reverse-phase chromatography using bonded phases and
methanol-water gradient
solvent systems, since these are more
compatible with the mass spectrometry steps [98]. This current study explored
other inexpensive methods such as colony assay, wound healing assay, and trans
well migration and invasion assay for the quantification of SC-514 release from
SC-514-PLGA nanoparticles.
Materials and Methods
Determination of drug solubility in
release media (10 mM phosphate buffered saline (pH 7.4) supplemented with 10%
(v/v) of FBS and 1% (v/v) PenStrep®
Prior to the release
experiments the thermodynamic solubility of SC-514 drug in release media was
tested. For this purpose, 100 mg the SC-514 drug was added to 3 mL of the
releasing medium and incubated at 37 °C for 24 hours. The release medium was
composed of a 10 mm phosphate buffered saline (pH 7.4) supplemented with 10%
(v/v) of FBS and 1% (v/v) Pen Strep® to avoid microbial growth. The mixture of
the SC-514 drug and release medium was collected in a micro centrifuge tube for
centrifugation. A solubility study was carried out by using centrifugation to
separate the particulate fraction of SC-514 drug in the release medium.
Conjugation of SC-514 loaded PLGA nanoparticle with NF-κB
antibody
10 µg of the NF-KB was added to 200 mg PLGA polymer for the Nano-formulation. The nanoparticles were formulated with
tween 80 as the surfactant optimizing the nanoparticles for NF-κB antibody ligand
conjugation. [99]. The final concentration of antibody in the NP solution was
approximately 0.06 µg/mL
Functionalization of SC-514-PLGA nanoparticles with fats and
oil
(melted animal fat was utilized) was carried out using
trimethylphenylammonium chloride (199168-100G) as cationic surfactant
(substances in which the hydrophilic, or water-loving, end contains a
positively-charged ion, or cation) ionically bonding the fats and oil to the
surface of the nanoparticles as adapted from previous studies [100-104].
Dialysis method of drug release
The effective drug concentration within the nanoparticle
provides the driving force for release from the particle in the release media
(phosphate buffered saline (pH 7.4) supplemented with 10% (v/v) of FBS). In-vitro release kinetics of
SC-514-PLGA, SC-514-PLGA-NF-KBAb, and SC-514-PLGA-Fat was investigated in this
study using dialysis bag method. Typically, SC-514-loaded nanoparticle suspension (1.0 mL) or drug solution
with the equivalent drug concentration was enclosed in a dialysis bag (MWCO 12
kDa) and then placed in 200 mL of pH 7.4 phosphate buffered saline solution
(supplemented with 10% (v/v) of FBS). The release medium was replaced with
fresh buffer every 24 hours. The entire system was kept at 37°C with continuous
magnetic stirring.
SC-514 PLGA Nanoparticles Instrument Settings

Table 1: LC (Shimadzu UFLC XR) conditions
Quantification of SC-514 released by LC–MS analysis of SC-514
PLGA nanoparticles
At 24 hours’ time intervals, 30 µL of aqueous solution was
withdrawn from the release medium and the SC-514 concentration was assayed
using ABSciex 5500 mass spectrometer. A standard curve was utilized to
determine the unknown quantity of SC-514 released in the aqueous solution. The
sample was put back to the release medium after the measurement. For
determining release kinetics of SC-514-PLGA suspension, a dialysis bag (12 kDa
MWCO) was used to enclose the sample (5 mL). The sealed dialysis bag was then
placed in a USP apparatus 2 containing 150 mL of pH 7.4 PBS at 37°C with a
paddle rotating at 100 rpm. At interval of 1 day for 30 days, 30 µL of release
medium was taken out and drug concentration was measured by HPLC and MS/LC. The
30 µl of each sample that was removed was added to 70 µl of acetonitrile
containing carbamazepine as the internal standard. Samples were compared to a
standard curve prepared in RPMI-1640 medium.
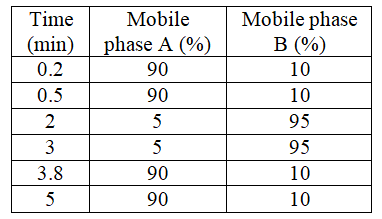
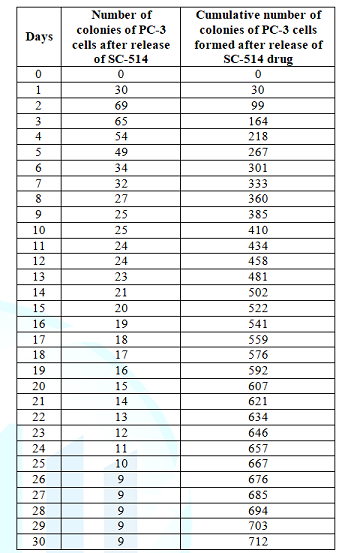
Table 4: PC-3 cells colonies counted for colony assay.
Alternative
methods for quantifying SC-514 drug released from SC-514 PLGA
The
results from colony forming assay are shown in Figure 5 and Table 4.
The number of days of drug release study and cumulative number of colonies of
PC-3 cells formed after release of SC-514 drug from SC-514-PLGA nanoparticles
was plotted to produce a drug release curve (Figure 3).
The results from the transwell
migration and invasion assay, shown in table 5 below are indicated in
the stained form (Figures 5, 6, and 7) and unstained form (Figure
8). The number of days of drug release study and number of PC-3
cells in the lower chamber of transwell after release of SC-514 drug from
SC-514-PLGA nanoparticles
was plotted to produce a drug release curve (Figure 3). As
drug release progressed from day 0 to day 30, number of PC-3 cells in
the lower chamber of transwell after release of SC-514 drug from SC-514-PLGA decreased.
However, as drug release progressed, cumulative number of PC-3 cells in the
lower chamber of transwell also increased.
The
results from the wound healing assay are indicated on day 1 to day 6 (Figure 9 and Table 6). The
number of days of drug release study and cumulative width over time after
release of SC-514 drug from SC-514-PLGA nanoparticles was plotted to produce a
drug release curve (Figure 3). As the drug release progressed from day 1 to day
30, the wound width created decreased from day 1 to day 30. However, the
cumulative wound width increased. Drug release curve was constructed for the
purpose of comparing different SC-514 quantification methods (Figure 3).

Table 5: PC-3 cells counted for transwell migration and invasion assay
Alternative
methods for SC-514 Drug Release Studies
Transwell assay staining of LNCaP
cells, PC-3 cells, and DU-145 cells that indicated a more consistent trend of
drug release pattern was observed in PC-3 cells than in LNCaP cells and DU-145
cells (Figure 5 and Figure 6).
The colonies formed from LNCaP cells were the most sensitive
to SC-514 drug release, followed by the colonies formed from DU-145 cells. The
colonies formed by PC-3 cells were the least sensitive to SC-514 drug release.
This trend is in consistency with aggressiveness of the prostate cancer cell
lines utilized (PC-3 cells are the most aggressive during proliferation and
LNCaP cells are the least aggressive). The higher the aggressiveness of the
prostate cancer lines, the lower the sensitivity of the colony cells formed to the SC-514 drug release.
Transwell assay staining of PC-3 cells suggested that more
SC-514 was released from SC-514-PLGA nanoparticles (cumulative release) as the
days progressed from day 1 to day 12, which correlated with the reduction in
number of PC-3 cells that migrated through the filter from day 1 to day 12.
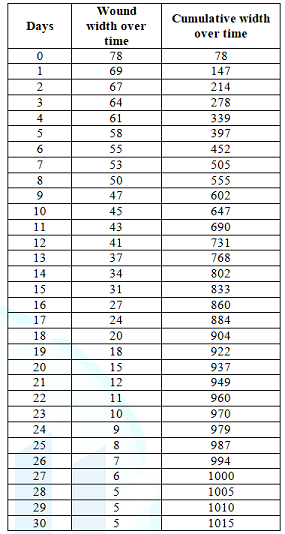
Table 6: Wound width between monolayer of PC-3 cells
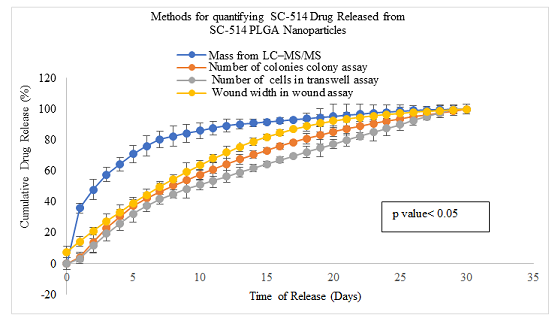
Figure 3: Four methodswere utilized to investigate the release of SC-514 drug from SC-514-PLGAnanoparticle over 30 days. Three of these methods were new and unconventional (Colony assay,transwell assay, and wound assay). Colony assay, transwell assay, and woundassay methods indicated a similar trend of drug release with no outburstrelease of the SC-514 drug. LC-MS/MS conventional method indicated that SC-514released from SC-514-PLGA encapsulations is a first order release curve withinitial outburst.
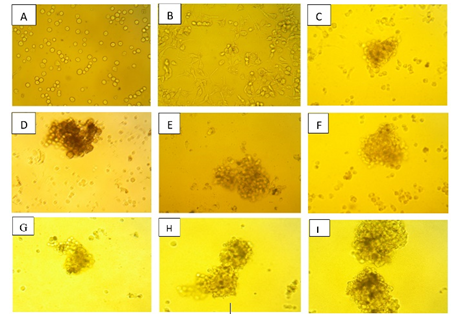
Figure 4: The result from colony forming assay ofPC-3 cells as an alternative method to quantify SC-514 drug release. A:0 h of cell culture, B: 48 h of control, C: release ofSC-514 from SC-514-PLGA on day 1, D: release of SC-514 from SC-514-PLGA on day2, E: release of SC-514 from SC-514-PLGA on day 3, F: release of SC-514from SC-514-PLGA on day 4, G: release of SC-514 from SC-514-PLGA on day 5, H:release of SC-514 from SC-514-PLGA on day 6, I: release of SC-514 from SC-514-PLGAon day 7.
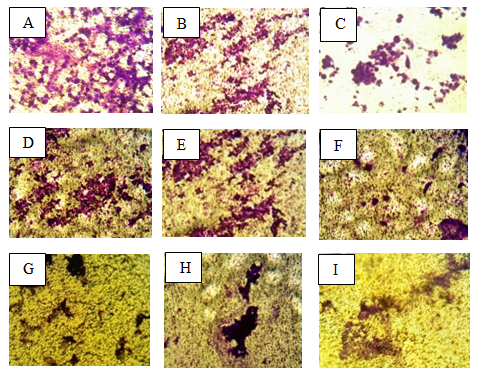
Figure 5: The result from colony forming assay of LNCaPcells, PC-3 cells, and DU-145 cells as an alternative method for SC-514 drugrelease study. The exper
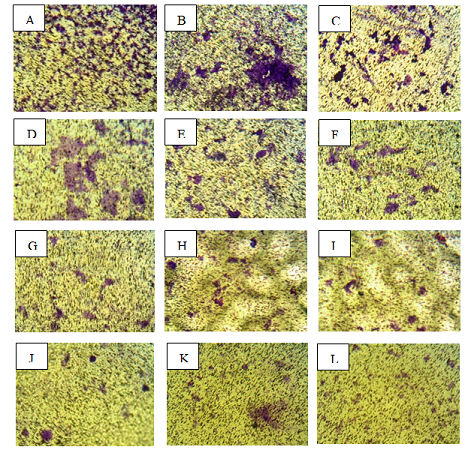
Figure 6: Transwell assay showing the number of PC-3 cells
Transwell assay showing the number of DU-145 cells that
migrated through the transwell after the release of SC-514 drug to the DU-145
cells from SC-514-PLGA nanoparticles.

Figure 7: Transwell assay showing the number of DU-145 cells
Transwell assay showing the number of unstained PC-3 cells
that migrated through the transwell after release of SC-514 drug from
SC-514-PLGA on the PC-3 prostate cancer cells from day 1 to day 30.

Figure 8: Transwell assay showingthe number of unstained PC-3 cells that migrated through the transwell afterrelease of SC-514 drug from SC-514-PLGA from day 1 to day 30. D1: release ofSC-514 drug from SC-514-PLGA on day1, D2: release of SC-514 drug from SC-514-PLGAon day 2, D3: release of SC-514 drug from SC-514-PLGA on day 3, D4: release ofSC-514 drug from SC-514-PLGA on day 4, D5: release of SC-514 drug fromSC-514-PLGA on day 5, D6: release of SC-514 drug from SC-514-PLGA on day 6, D7:release of SC-514 drug from SC-514-PLGA on day 7, D8: release of SC-514 drugfrom SC-514-PLGA on day 8, D9: release of SC-514 drug from SC-514-PLGA on day9, D10: release of SC-514 drug from SC-514-PLGA on day 10, D11: release ofSC-514 drug from SC-514-PLGA on day 11, D12: release of SC-514 drug fromSC-514-PLGA on day 12, D13: release of SC-514 drug from SC-514-PLGA on day 13,D14: release of SC-514 drug from SC-514-PLGA on day 14, D15: release of SC-514drug from SC-514-PLGA on day 15, D16: release of SC-514 drug from SC-514-PLGAon day 16, D17: release of SC-514 drug from SC-514-PLGA on day 17, D18: releaseof SC-514 drug from SC-514-PLGA on day 18, D19: release of SC-514 drug fromSC-514-PLGA on day 19, D20: release of SC-514 drug from SC-514-PLGA on day 20,D21: release of SC-514 drug from SC-514-PLGA on day 21, D22: release of SC-514drug from SC-514-PLGA on day 22, D23: release of SC-514 drug from SC-514-PLGAon day 23, D24: release of SC-514 drug from SC-514-PLGA on day 24, D25: releaseof SC-514 drug from SC-514-PLGA on day 25, D26: release of SC-514 drug fromSC-514-PLGA on day 26, D27: release of SC-514 drug from SC-514-PLGA on day 27,D28: release of SC-514 drug from SC-514-PLGA on day 28, D29: release of SC-514drug from SC-514-PLGA on day 29, D30: release of SC-514 drug from SC-514-PLGAon day 30.
Wound healing assay of unstained PC-3 cells was conducted as
an alternative method for drug release study. On day 6 of drug release (Figure 9H), there were less
fibroblastic PC-3 cells compared to the day 6 control with no release of SC-514
drug from SC-514-PLGA (Figure 9I).
Also, the PC-3 cells in the wells that received cumulative release of SC-514
drug were rounded up, clumped together and not well attached to the surface of
the culture plate by day 6 (Figure 9H). On the other hand, the PC-3 cells with
no release of SC-514 drug from SC-514-PLGA (control) on day 6 appeared
elongated, spaced out at good distance and well-attached the surface of the
wells (Figure 9I).

Figure 9. Wound healing assay of unstained PC-3 cells
The intracellular delivery of SC-514 from poly (lactide-co-glycolide)
(PLGA) nanoparticles stabilized with bovine serum albumin, in PC-3 cells, was
studied via confocal microscopy (Nikon A1R Confocal System w/SIM). As the
incubation time changes, florescence intensity and cellular uptake changes (Figure 11).
The cellular uptake efficiency of nanoparticles in PC-3
prostate cancer cell was higher in the SC-514-PLGA-NF-KBAb NPs than SC-514-PLGA
NPs (Figure 13). This is consistent
with the results of the drug release study (Figure 2) and the impact of SC-514
nanoparticle formulations on PC-3 cells (Figure
19 and 20) and cord blood cells (Figure 17 and figure 18). It takes a
longer time for SC-514-PLGA-NF-KBAb to release the SC-514 drug content because
of the high cellular uptake efficiency of the whole nanoparticles in cells. On
the other hand, SC-514-PLGA NPs has lower cellular uptake efficiency with a
burst release at the beginning of the drug release study.
The degree of enhanced cellular accumulation of PLGA-SC-514
NPs was higher in prostate cancer cells than cord blood cells (Figure 12). The underlying mechanisms
of enhanced cellular accumulation efficiency of SC-514-PLGA NPs compared with
that of PLGA NPs should be further investigated. Generally, there was an
increased concentration of nanoparticles in PC-3 cells because of increased
incubation time.
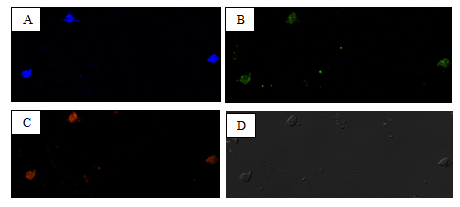
Figure10: Picture showing PC-3 cells and SC-514-PLGAnanoparticles. A: Nucleus stain of PC-3 cells with DAPIB: Expression of multidrug resistance in PC-3 cells, C: SC-514-PLGAnanoparticles in PC-3 cells, D: Phase contrast image of PC-3 cells.These images represent x-y confocal images (20x magnification).
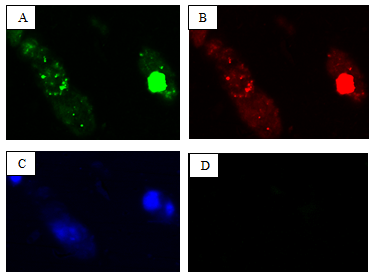
Figure 11: Confocal microscopy ofPC-3 prostate
The degree of cellular accumulation of PLGA-SC-514 NPs was
higher in prostate cancer cells than cord blood cells (Figure 12). The effect of the nanoparticle treatment on PC-3 cells
was also investigated (Figure 14).
This study utilized immunofluorescence assay to investigate the expressing of
MDR proteins after treatment with free SC-514 and SC-514-PLGA nanoparticles. SC-514-PLGA nanoparticles reduced the
expression of MDR protein in PC-3 cells significantly more than free SC-514. Controlled
and optimum delivery of SC-514 drug from the nanoparticle treatment PLGA NPs
has the potential to eliminate the imbalance in the length of drug treatment
favoring MDR in prostate cancer. This will potentially reduce the expression of
P-gp and other ABC transporter proteins in prostate cancer during treatment. Reduction
in cell viability was observed when PC-3 cells were incubated with the
nanoparticle formulations for 48 h at 37° C and 5% CO2. Varying
concentrations of SC-514 released from the nanoparticle formulation inhibited
the cell growth. The growth inhibition was manifested by shrinking and
granulation of PC-3 prostate cancer cells (Figure
15).

Figure12: The degree of cellular accumulation of PLGA-SC-514NPs was higher in prostate cancer cells than cord blood cells. The result is based onthe intensity of color at any point in time. A: The Green fluorescence indicated cellular accumulation of PLGA-SC-514 NPs in PC-3 cells,B: The red fluorescence indicated cellular accumulation of PLGA-SC-514 NPs in cord bloodcells.
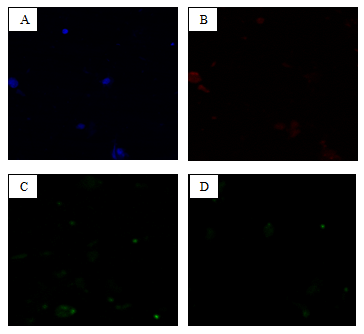
Figure 13: Quantitative study of PLGA nanoparticles uptake in PC-3 cells
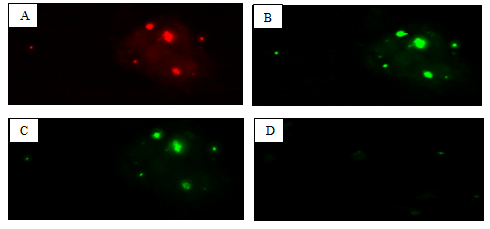
Figure 14: Expression of MDR in PC-3 prostate cancer cells. A: SC-PLGA
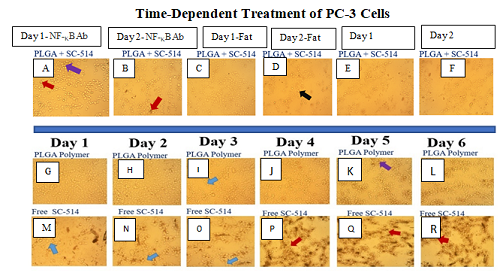
Figure 15: The appearance and structural characteristics of PC-3 prostate cancer cells
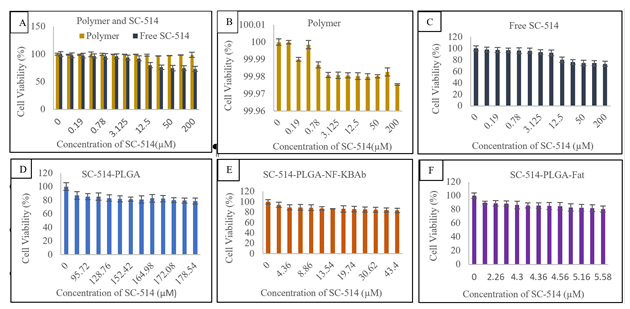
Figure16: Cord blood cells treatment with SC-514 drug releasefrom nanoparticle treatments at different concentrations impacting differentlevels of cell viabilities.A:comparison between cell viabilities of cord blood cells treated with polymerand free SC-514, B: cell viability of cord blood cells treated with polymer, C:cell viability of cord blood cells treated with free SC-514, D: cell viabilityof cord blood cells treated with SC-514-PLGA, E: cell viability of cord bloodcells treated with SC-514-PLGA- NF-KBAb, F: cell viability of cordblood cells treated with SC-514-PLGA-Fat.

Figure17: Cord blood cells treatment with SC-514 drug releasefrom nanoparticle treatments impacted different levels of cell viability fromday 1 to day 12. Concentrations ofpolymer and free SC-514 from day 1 to day 12 ranged from 0 µM to 200 µM.Concentrations of the other nanoparticle treatments (SC-514-PLGA,SC-514-PLGA-NF-KBAb, SC-514
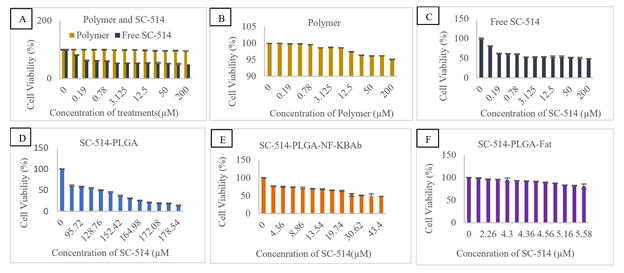
Figure 18: PC-3 cells treatment with SC-514 drug
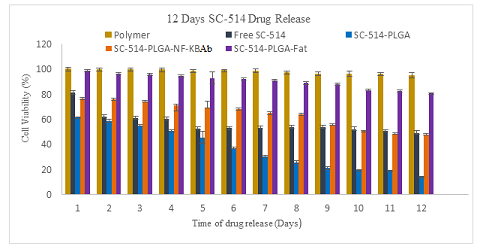
Figure 19: The cell viability of PC-3cells after treatment with SC-514 drug concentrations obtained from the in-vitro drug relea
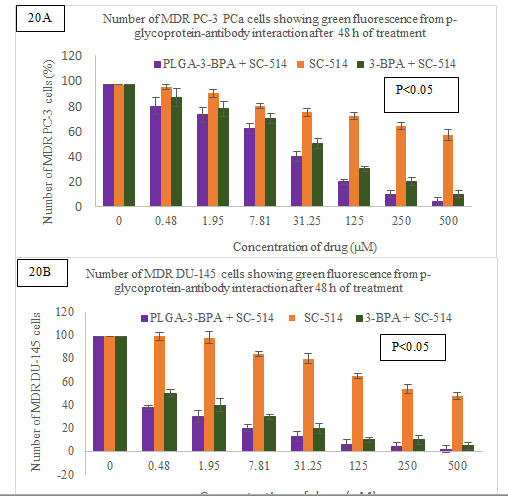
Figure 20: Immunofluorescence analysis results detecting
Result and Discussion
The conventional cancer
chemotherapy has many negative effects such as Multiple Drug Resistance (MDR),
high clearance rate (pharmacokinetic measurement of the volume of plasma from
which a drug is completely removed per unit time), severe side effects,
unwanted drug distribution to the normal cells and low concentration
of drug at the site of prostate cancer cells [111]. Therefore, it is necessary
to develop novel strategies and novel Nano carriers that will carry the drug
molecules directly to the affected cancerous cells in an adequate amount and
duration within effective therapeutic window [112,113]. Nanoparticle drug
delivery systems have advantages over conventional chemotherapy due to the high
efficacy of drug loading or drug encapsulation efficiency, high cellular
uptake, high drug release, and minimum side effects. These Nano carriers
possess high drug accumulation in the tumor area while minimizing toxic effects
on healthy prostate tissues [112].
To reduce MDR in prostate cancer
treatment, this study investigated the therapeutic advantage of encapsulating
SC-514 in PLGA polymer and conjugating the surface of the nanoparticles formed
to further control drug delivery. The water-insoluble SC-514 drug in a
hydrophobic PLGA based matrix showed average drug loading due to leaching
effects (uncontrolled accidental release of drug). Therefore, SC-514-PLGA nanoparticles
were conjugated with NF-KB antibody and Fats. It was necessary to develop a
useful method to increase the drug encapsulation efficiency and improve the
drug bioavailability of SC-514.
Further improvement of SC-514
drug entrapment by conjugation during nanoparticle formulation can be
considered advantageous in reduction of multidrug resistance in prostate
cancer. This is important because prolonged drug release has been shown to
reduce drug resistance in cancer treatment [114,115]. The goal of this study
was to investigate SC-514 drug release from nanoparticle formulations that has
the potential to reduce multidrug resistance by sustained release of SC-514
drug from the PLGA nanoparticle formulations.
The use of Nano
encapsulation of SC-514 will improve the chance to
target the prostate cancer cells and not harm normal prostate cells because
targeted drug delivery of nanoparticles decorated with site-specific
recognition ligands is of considerable interest to minimize cytotoxicity of
chemotherapeutics in the normal cells [115]. SC-514 was internalized into the
PLGA nanoparticles by endocytosis which may be released via endosome escape
delivering the encapsulated SC-514 drug to the cytosol of the cells. Higher
intracellular delivery of the SC-514 drug from the nanoparticles suggested a
high efficacy of encapsulated SC-514 drug. Hence, lower dose of the SC-514
nanoparticle formulation could produce a higher cytotoxic effect on the cancer
cells than free SC-514 drug (Figure 16 and Figure 18). Previously,
SC-514-loaded poly (lactic-co-glycolic acid) (PLGA) nanoparticles (SC-514-PLGA)
were prepared by the single emulsion method. The influence of different
experimental parameters on the incorporation of SC-514 in the nanoparticles was
evaluated. Functionalized SC-514 PLGA nanoparticles were prepared based on
previously modified method [46]. We utilized various techniques for drug
solubility enhancement including nanoparticle surface functionalization. The
surface of SC-514-PLGA polymeric drug delivery system was functionalized with NF-KB
antibody and fat to form SC-514-PLGA-NF-KBAb and SC-514-PLGA-Fat respectively.
The functionalization was done to further improve the therapeutic index of
SC-514 drug and reduce the adverse treatment effects in this current study.
This impact of of functionalization in this current study is similar the
results from other studies [116,117].
Delivering chemotherapeutics by
nanoparticles into a tumor
is mostly impeded by two factors: nonspecific targeting and inefficient
penetration. Targeted delivery of anti-cancer agents solely to tumor cells
introduces a smart strategy because it enhances the therapeutic index compared
to untargeted mode of delivery of drugs [118]. The anti-cancer effect of SC-514
nanoparticle formulations (SC-514-PLGA, SC-514-PLGA-NF-KBAb, and
SC-514-PLGA-Fat nanoparticles) on cord blood cells and PC-3 cells was
investigated.
The release behavior of SC-514
from the developed SC-514-PLGA exhibited a biphasic pattern characterized by an
initial fast release during the first 24 hours, followed by a slower and
continuous release (Figure 2). This is very similar to the burst release
observed in other studies [119-121], thus, confirming the treatment efficacy of
the nanoparticle
delivery approach. Drug release from SC-514-PLGA
nanoparticles appears to consist of two components with an initial rapid
release followed by a slower exponential stage (Figure 2). SC-514-PLGA
nanoparticles indicated that 50% of the drug was released on the 3rd day. The
SC-514-PLGA-NF-KBAb indicated that 50% of the drug was released on the 20th
day. SC-514-PLGA-Fat indicated that 50% of the drug was released on the 12th
day. For all the nanoparticle formulations, 50% of drug release extended from
hours to weeks in this current study.
A model predicts a two-stage
release profile, with a relatively rapid initial release of most of the drug,
followed by a slower release of the remaining drug known as a “plateau” phase [122].
This is consistent with the results from SC-514-PLGA drug release in this study
(Figure 2). Faster initial release of SC-514 in SC-514-PLGA than in
SC-514-PLGA-NF-KBAb and SC-514-PLGA-Fat might be due to a faster dissociation
of polylactic acid-glycolic acid polymer.
Generally, low-MW drugs,
peptides, and proteins have higher propensities for burst release as a result
of osmotic pressures [123].
In most treatments, a strong
burst release is to be avoided as it decreases the efficacy of the treatment
and can be dangerous to the host [120]. It may also waste the SC-514 drug if
the excess drug cannot be absorbed by the body within the time of
administration. Although under certain circumstances an initial sharp release
of the therapeutic agent could be desirable, it is often unpredictable with
uncontrollable duration and dose [8,123]. For example, if a sudden voluminous
delivery is required, burst release can be triggered by rapid changes in the
local environment. However, for the most part, avoiding the burst release
effect is desirable to minimize any initial toxicity associated with a high
dose. For this reason, various methods have been recommended to control
unnecessary and dangerous burst release of drugs [123] as seen with the
SC-514-PLGA nanoparticles drug release in this current study. To maximize the
effectiveness of nanoparticle targeting, drug release from nanoparticles needs
to be slow enough to avoid substantial drug loss before the carrier reaches the
site of action thereby reducing toxicity [124,125]. Initial burst can be
further controlled by modifying the solidification
rate of the dispersed phase [126]. In order to prevent many unfavorable events
such as pore formation, drug loss, and drug migration that occurred while the
dispersed phase is in the semi-solid state, it is important to understand and
optimize the nanoparticle formulation variables [127]. In this study, we
functionalized the surface of PLGA nanoparticles with molecules including NF-KB
antibody and fat to remove the burst release problem observed previously.
SC-514-PLGA was conjugated with
NF-KB antibody in order to investigate the impact of NF-KB antibody rich
microenvironment on SC-514 drug release. Elevated expression of NF-KB has been
implicated in prostate cancer carcinogenesis [128]. Also, the high expression
of NF-κB within the cancer cells might be used for the eradication of selective
cancer cells that could be regulated by the modulation of the NF-κB pathway [129].
Viable prostate cancer cells
increase NF-κB translocation to the nucleus with subsequent enhancement of bot h activation of NF-κB transcription and
induction of NF-κB responsive genes. This increase in NF-κB expression may be
related to the NF-κB function as a transcription factor, which can explain the
increase of cancer
cells division [130]. NF-κB is highly
expressed in actively proliferating prostate cancer. NF-κB provides surface
accessibility and preferred accumulation of antibody-conjugated Nano carriers
through receptor-mediated endocytosis [131]. Conjugating a nanoparticle with
appropriate surface molecules such as NF-κB may trigger and control drug
release properties and prolong drug release time [132]. The results from the
conjugation experiment involving NF-κB and SC-514 demonstrated the
possibilities of modulating the release profile by means of modifying the
surface of the nanoparticles for higher encapsulation efficiency consistent
with other studies. [133-136]. Drug delivery systems with high drug
encapsulation efficiency and controlled release are of great importance in
biomedical fields [137].
In addition, some membrane
transport proteins maybe implicated in the endocytosis of PLGA nanoparticles in
prostate cancer cells. These membrane transport proteins may play a role in
PLGA nanoparticle endocytosis [138] in PC-3 and cord blood cells. The plasma
membrane can be crossed by PLGA NPs with a
diameter of 500-600 nm [139]. It appeared that the difference in nanoparticle
sizes may cause the difference in nanoparticle retention between
SC-514-PLGA-NF-κBAb nanoparticles and SC-514-PLGA nanoparticles. Physical
characterization showed that the antibody unconjugated and conjugated particles
were oval to spherical and within the size range of 200–250 nm. This current
study indicated that the average particle size slightly increased for
antibody-conjugated nanoparticles SC-514-PLGA-NF-κBAb compared to the
SC-514-PLGA. This implies that a hard decision needs to be made based on the
preference for particle size and the length of time it takes to release SC-514
drug.
The SC-514 release from SC-514
PLGA nanoparticles was investigated using dialysis method. The principle was
based on a change in the permeation rate of the small molecule across the
dialysis membrane with the change in the free fraction (or fraction bound)
inside the dialysis chamber [140,141]. The application of the dynamic dialysis
method for determining release kinetics from nanoparticles seems to have grown
in popularity, in part due to the willingness of investigators to ignore the
demerits of dynamic dialysis method [142]. With the ever-increasing research
efforts in the field of nanoparticles as drug
delivery systems, it is critical to understand
the limitations of this widely adopted dynamic dialysis method for
determination of release kinetics. There are various scenarios where the
interpretation of release data using dialysis can be either inaccurate or
completely misleading. As shown in this study, consideration of the binding
affinity of the drug to the nanoparticles, appropriate control experiments, and
suitable mechanism-based mathematical treatment of the data should aid in the
judicious use of the dialysis method for determination of the release kinetics
from nanoparticles [143].
The dual barrier nature inherent
in the dynamic dialysis method complicates data interpretation and may lead to
incorrect conclusions regarding nanoparticle release half-lives. Although the
need to consider the barrier properties of the dialysis membrane has long been
recognized, there is an insufficient quantitative appreciation for the role of
the driving force for drug transport across that membrane. Reversible Nano
carrier binding of the released drug reduces the driving force for drug
transport across the dialysis membrane leading to a slower overall apparent
release rate [144]. This may lead to the conclusion that a given nanoparticle
system will provide a sustained release in-vivo.
However, this not always true.
Although the equilibrium dialysis
method can achieve separation of nanoparticles from the surrounding solution,
this method can produce misleading in-vitro
release data. To date, no standardized
technique for the assessment of drug release from
Nano medicines has been issued by regulatory authorities. In view of the
shortcomings of dialysis methods, pressure ultrafiltration has been proposed as
an alternative dialysis method that can produce a release profile that is
representative of the true distribution of the drug between the nanoparticle
and the dispersing medium at any point in time [145].
A potential issue associated with
the use of any of the physical separation methods is that the separation may be
incomplete or inefficient. It is impossible to visually detect the presence of
a small number of nanoparticles present in the filtrate or supernatant of a
separated sample. However, their presence is likely to lead to significant
measurement errors, particularly early in the release timescale when the
concentration of drug in the carrier particles is high relative to that free in
solution. The application of such separation methods is frequently reported in
the drug delivery literature, however to our knowledge there has been no method
proposed to validate the efficiency of separation of nanoparticles from the
surrounding medium in which they are dispersed to produce a ‘clean’ sample of
unbound drug. The human body is able to adapt to a little inefficiency between nanoparticles
and surrounding medium because a research showed that a large amount of PLGA
nanoparticles were present in the kidney and liver, without causing any
morphological changes in respective tissues, even at a high dose of PLGA. This
emphasizes the fact that PLGA nanoparticles are safe in the kidney and liver
when they are used to deliver any incorporated drug [146].
The US-FDA has also recommended
it as nontoxic and safe for human use. There is no report of its toxic effect
on the kidney and liver [147]. This is consistent with the results from this
study. PLGA polymer impacted the lowest amount of toxic effect on cord blood
cells, followed by SC-514-PLGA-NF-κBAb, SC-514-PLGA-Fat, SC-514-PLGA, and then
free SC-514 (Figure 16 and Figure 17). PLGA enhanced therapeutic potency even
at low concentration of SC-514 drug released.
In-vivo
treatment of PC-3 cells in a mouse model with
SC-514-PLGA-NF-κBAb will potentially support the site-specific drug delivery
ability of the formulation and therapeutic potential of formulated Nano
carriers in the treatment of NF-κB -overexpressed prostate cancers. In this
study, antibody-conjugated SC-514-loaded PLGA nanoparticles showed a promise in
improving the tumor site-specific delivery of the drug with a significant
reduction of drug-related toxicity (Figure 16 and Figure 17). SC-514-PLGA
showed therapeutic improvement over free-SC-514, SC-514-PLGA-NF-κBAb and
SC-514-PLGA-Fat on the first day of drug release to PC-3 prostate cancer cells
(Figure 18 and Figure 19). However, SC-514 drug release from
SC-514-PLGA-NF-κBAb and SC-514-PLGA-Fat may be advantageous for prolonged and
sustained drug release needed to reduce MDR in prostate cancer. Hence,
SC-514-PLGA-Fat or SC-514-PLGA-NF-κBAb could be a preferential choice to
deliver SC-514 drug more specifically in MDR-overexpressed prostate cancer
cells.
During the first 7 days of
cumulative drug release, free SC-514 had lower toxicity (most likely because of
low solubility) than SC-514-PLGA-NF-κBAb. However, after 7 days the toxicity of
free SC-514 was higher than the toxicity of SC-514-PLGA-NF-κBAb (Figure 17).
This is consistent with the results from our previous study that indicated
higher anti-cancer activities for SC-514 at high concentrations [46].
The evaluation of the side
effects of the nanoparticle systems compared to the free SC-514 may indicate
apparent similar side effects based on cell viability study with cord blood
cells (Figure 16 and Figure 17). However, a prolonged sustained release of
SC-514 drug from SC-514-PLGA, SC-514-PLGA-NF-κBAb and SC-514-PLGA-Fat has
therapeutic advantage to overcome MDR in prostate cancer. Specifically,
PLGA-3-BPA + SC-514 nanoparticle treatment reduced the number of MDR PC-3 cells
and MDR DU-145 cells significantly when compared to SC-514 and 3-BPA +SC-514
treatments.
The nanoparticle formulations
have the potential to preferentially deliver SC-514 drug to the tumorigenic
cells, causing reduction of SC-514 mediated toxicity due to its more
site-specific distribution of drug to the target site. Further, due to
sustained and controlled drug release from the formulation, much less free drug
will reach the heart and other parts of body tissue to cause cardiac toxicity
and systemic toxicity respectively. Thus, this formulation may offer future
hope to deliver the drug to the target cancer tissue and minimize toxicity
of the drug to normal tissue. However, the major limitation of this
antibody-conjugated formulation such as SC-514-PLGA-NF-κBAb is the saturation
of cell surface target protein (antigen). Once the surface antigen proteins are
saturated, the formulation would not be able to target the neoplastic cells
only and prolong presence without its distribution in neoplastic cells, which
may affect normal prostate cells [115].
Thus, the dose of
SC-514-PLGA-NF-κBAb nanoparticle should be optimized before administration of
formulation in-vivo. Hence, further
studies including animal model studies and clinical trials are needed to
optimize the dose of SC-514 in human subjects and to investigate the clinical
efficacy of the formulation in the human prostate cancer patients. Efficient
quantification of SC-514 drug could support optimization of SC-514 drug release
for in-vitro and in-vivo studies. In this study, high-performance liquid
chromatography (HPLC), LC/mass spectrometry (MS) and LC/tandem mass
spectrometry (MS/MS) were utilized as the standard method to quantify SC-514
drug released from SC-514-PLGA nanoparticles. HPLC and LC/MS have been widely
used for biomedical
analyses, in which chemical derivatization (a
technique used in chemistry which converts a chemical compound into a product
of similar chemical structure) is one of the most important methods to increase
sensitivity and selectivity [148,149].
LC-MS/MS offers improved levels
of accuracy and reproducibility over traditional methods. LC-MS/MS has emerged
as the latest technology utilized for drug release studies. However, this
technology is not readily available to most researchers [150]. There is a need
to investigate new methods for drug release studies. In this study, we
investigated the use of other methods such as colony forming assay, transwell
invasion and migration assay, and wound healing assay as alternative methods
for drug release studies.
During the transwell assay,
necessary precaution was taken to avoid washing off fixed cells from the
membrane. The cell dilutions were worked out and the dishes were labeled
appropriately. The experiment was conducted continuously to limit the total
time, preventing adverse effects of pH and temperature changes. It is important
to note that there are distinct differences between the transwell cell
migration and the transwell cell invasion assays. The transwell cell migration
assay measures the chemotactic
capability of cells toward a chemo-attractant. The
transwell cell invasion assay, however, measures both cell chemo taxis and the
invasion of cells through extracellular matrix, a process that is commonly
found in prostate cancer metastasis. In this study we utilized both transwell
cell migration and invasion assay as an alternative method for LC/MS. The
number of PC-3 cells that migrated was counted manually using a counter. Other
studies utilized I-AbACUS, a software tool specifically designed to aid the
analysis of the transwell assays that automatically and specifically recognized
cells in images of stained membranes and provided the user with a suggested
cell count.
Comparison between I-AbACUS and
the standard technique for analysis of the transwell assay indicated that the
manual count had an average error below 10%. Although transwell and invasion
migration assay, colony forming assay, and wound healing assay are techniques
that have been used extensively in multiple research studies [151-154]. This
current study is the first study that explored transwell and invasion migration
assay, colony forming assay, and wound healing assay as a method of quantifying
drug release.
The three alternative methods of
quantifying SC-514 drug released from SC-514-PLGA nanoparticles discussed above
did not show burst release like the LC-MS method. The LC-MS method consistently
indicated the highest cumulative SC-514 drug released (Figure 3). The pattern
of drug release was similar for all the three alternative methods without burst
release (wound healing assay, colony
forming assay, and transwell migration and invasion
assays). The wound assay consistently indicated higher level of cumulative
release compared to colony assay method and transwell method. Between day 9 and
day 23 of drug release, there was a clear difference between the cumulative
release levels of the three alternative methods: the highest release was
observed in the wound assay method, followed by the colony assay then the
transwell assay (Figure 3).
Dissolution
of a drug is the rate determining step for oral absorption of the poorly
water-soluble drugs and solubility is the basic requirement for the absorption
of the drug. A proper selection of solubility enhancement method is the key to
ensure the goals of a good formulation like good oral bioavailability, reduce
dosage frequency and better patient compliance combined with a low cost of
production. Selection of methods for solubility enhancement depends upon drug
characteristics like solubility, chemical nature, melting point, absorption
site, physical nature, pharmacokinetic behavior, dosage form requirement like
tablet or capsule formulation, strength, immediate, or modified release, and
regulatory requirements like maximum daily dose of any excipients and/or drug,
approved excipients, and analytical accuracy [45].
although a reported study
indicated that endocytosis of nanoparticles in primary cultured RCECs occurred
mostly independent of clathrin- and caveolin-1-mediated pathways, other
proteins maybe involved in the endocytosis of PLGA nanoparticles in the PC-3
cells and cord blood cells. The internalization of poly
(dl-lactide-co-glycolide, PLGA) nanoparticles in prostate cancer cells occurred
by an endocytic process, regulated by availability of energy [138]. Inhibition
of ATP energy in prostate
cancer cells is expected to regulate the
internalization of PLGA nanoparticles by the cells. Fluorescent cell uptake
corroborated the receptor mediated endocytosis pathway, indicating the role of
adenosine receptors in internalization of conjugated particles. This internalization
was observed under confocal microscopy (Nikon A1R Confocal System w/SIM).
The higher uptake of the
nanoparticles by prostate cancer cells than cord blood cells was confirmed with
confocal microscopy (Figure 12). Cellular uptake of SC-514 was time dependent
and occurred potentially via endocytosis mechanism. This study tested prostate
cancer cells in-vitro with
traditional free SC-514 in comparison with poly lactic co-glycolic acid
nanoparticles carrying SC-514 (SC-514-PLGA, SC-514-PLGA-NF-κBAb, and
SC-514-PLGA-Fat).
Although, PLGA was conjugated
with encapsulating parthenolide, a NF-κB inhibitor, in order to improve the
selectivity and targeting of cancer cells while protecting the normal cells [155],
this current study appears to be the first study to functionalize the surface
of PLGA with NF-κB antibody or fats.
There is a high probability that
NF-κB-conjugated PLGA nanoparticles and fat-conjugated PLGA nanoparticles
containing SC-514 preferentially delivered encapsulated SC-514 drug to the
prostate cancer cells. This site-specific delivery of the formulation to
neoplastic cells would have minimal toxic effect on normal cells such as
prostate cells and white blood cells.
The ligand conjugated
nanoparticles further showed considerable potential in reduction of toxicity,
a prominent side-effect of the drug. Since conjugation increases the size of
nanoparticles [156] and smaller particles with large surface area are more
soluble than larger particles with smaller surface area [45] that means
SC-514-PLGA-NF-κBAb and SC-514-PLGA-Fat nanoparticles will have a lower
solubility than SC-514-PLGA because SC-514-PLGA- NF-κBAb and SC-514-PLGA-Fat
nanoparticles were larger in size. The major consideration will be to determine
whether increase solubility of SC-514 is more important than controlled
prolonged drug release of SC-514 for multidrug resistance reduction in prostate
cancer treatment.
Functionalized NPs reduce
toxicity and side effects of drugs. Also, functionalize NP support crossing the
biological barriers, such as the blood–brain barrier, and different cellular
compartments, including the nucleus [157]. Functionalization enhances the
properties and characteristics of nanoparticles through surface modification;
and enables them to play a major role in the field of medicine. Nanoparticle
drug delivery could be a promising new approach for personalized medicine. The
optimized formulation was covalently conjugated to NF-κB antibody and fats and
oils. Surface conjugation of the ligand was assessed by confocal microscopy.
Selectivity and cytotoxicity of
the experimental nanoparticles were tested on human prostate cancer and cord
blood cells utilizing MTT assay. The NF-κB -conjugated and unconjugated
nanoparticles were examined under a confocal microscope. In this study, we
utilized confocal microscopy to investigate functionalized nanoparticles. Other
techniques have been employed to investigate the functionalized NPs, including
exclusion chromatography (SEC).
The properties of PLGA
carrier-cargo system and release might be strongly influenced by the
combination of factors, including the individual properties of loaded compounds,
surface modification of the nanoparticles, and microenvironment. Thus, it is
unlikely that a single nanoparticle formulation will be identified that is
universally effective for the delivery of different compounds. The performance
of anti-cancer agents used in cancer diagnoses and therapies are improved by
enhanced cellular internalization of smart Nano carriers and controlled drug
release. In this study, SC-514-PLGA- NF-κBAb nanoparticles improved the
bioavailability and selective targeting of prostate cancer cells compared to
free SC-514, thus holding promise as a drug delivery system to improve the cure
rate of prostate cancer.
Conclusion
The results from this study will
encourage the development of drug
delivery systems for the local delivery of
anti-cancer drugs. PLGA drug delivery system will be advantageous to decrease
the concentration of administered SC-514 drug and the frequency of
administration, and subsequently minimizing the adverse effects that are faced
by prostate cancer patient during treatment. Findings from this study will
contribute to the rational design of other drug delivery systems with high drug
encapsulation efficiency and controlled release for treatment of various
cancers.
References
- Park TG. Degradation of poly (lactic-co-glycolic acid) microspheres: effect of copolymer composition (1995) Biomaterials 16: 1123-1130. https://doi.org/10.1016/0142-9612(95)93575-X
- Jain RA. The manufacturing techniques of various drug loaded biodegradable poly (lactide-co-glycolide) (plga) devices (2000) Biomaterials 21: 2475-2490. https://doi.org/10.1016/S0142-9612(00)00115-0
- Sahoo SK, Panyam J, Prabha S and Labhasetwar V. Residual polyvinyl alcohol associated with poly (d,l-lactide-co-glycolide) nanoparticles affects their physical properties and cellular uptake (2002) Journal of Controlled Release 82: 105-114. https://doi.org/10.1016/S0168-3659(02)00127-X
- HansML and Lowman AM. Biodegradable nanoparticles for drug delivery and targeting (2002) Curr Opin Solid State Mater Sci 6: 319-327. https://doi.org/10.1016/S1359-0286(02)00117-1.
- Yingchoncharoen P, Kalinowski DS and Richardson DR. Lipid-based drug delivery systems in cancer therapy: what is available and what is yet to come (2016) Pharmacological Reviews 68: 701-787. https://doi.org/10.1124/pr.115.012070
- Ulbrich K, Holá K, Šubr V, Bakandritsos A, Tuček J, et al. Targeted drug delivery with polymers and magnetic nanoparticles: covalent and noncovalent approaches, release control, and clinical studies (2016) Chemical Reviews 116: 5338-5431. https://doi.org/10.1021/acs.chemrev.5b00589
- Siegal T. Which drug or drug delivery system can change clinical practice for brain tumor therapy? (2013) Neuro-Oncology 15: 656-669. https://doi.org/10.1093/neuonc/not016
- Kamaly N, Yameen B, Wu J and Farokhzadm OC. Degradable controlled-release polymers and polymeric nanoparticles: mechanisms of controlling drug release (2016) Chemical Reviews 116: 2602-2663. https://doi.org/10.1021/acs.chemrev.5b00346
- Nochos A, Douroumis D and Bouropoulos N. In-vitro release of bovine serum albumin from alginate/hpmc hydrogel beads (2008) Carbohydrate Polymers 74: 451-457. https://doi.org/10.1016/j.carbpol.2008.03.020
- Makadia HK and Siegel SJ. poly lactic-co-glycolic acid (plga) as biodegradable controlled drug delivery carrier (2011) Polymers 3: 1377-1397. https://doi.org/10.3390/polym3031377
- Chereddy KK, Vandermeulen G and Préat V. PLGA based drug delivery systems: Promising carriers for wound healing activity (2016) Wound Repair and Regeneration 24: 223-236. https://doi.org/10.1111/wrr.12404
- Zhang E, Zhukova V, Semyonkin A, Osipova N, Malinovskaya Y, et al. Release kinetics of fluorescent dyes from PLGA nanoparticles in retinal blood vessels: In-vivo monitoring and ex vivo localization (2020) European Journal of Pharmaceutics and Biopharmaceutics 150: 131-142. https://doi.org/10.1016/j.ejpb.2020.03.006
- Bobo D, Robinson KJ, Islam J, Thurecht KJ and Corrie SR. Nanoparticle-based medicines: a review of fda-approved materials and clinical trials to date (2016) Pharmaceutical Research. https://doi.org/10.1007/s11095-016-1958-5
- Lü JM, Wang X, Marin-Muller C, Wang H, Lin PH, et al. Current advances in research and clinical applications of plga-based nanotechnology (2009) Expert Review of Molecular Diagnostics 9: 325-341. https://doi.org/10.1586/erm.09.15
- Shi J, Kantoff PW, Wooster R and Farokhzad OC. Cancer nanomedicine: Progress, challenges and opportunities (2017) Nature Reviews Cancer 17: 20-37. https://doi.org/10.1038/nrc.2016.108
- Welt FGP and Edelman ER. Adv Drug Delivery Rev Cell cycle regulation and control of angioplasty restenosis (1997) Advanced Drug Delivery Reviews 25: 299 https://doi.org/10.1016/s0169-409x(97)90003-x
- Y, Wu X, Mi Y, Zhang B, Gu S, et al. PLGA nanoparticles for the oral delivery of nuciferine: Preparation, physicochemical characterization and in-vitro/in-vivo studies (2017) Drug Delivery 24: 443-451.https://doi.org/10.1080/10717544.2016.1261381
- Duncan R. Nanomedicine gets clinical (2005) Materials Today 8: 16-17. https://doi.org/10.1016/S1369-7021(05)71032-4
- Brigger I, Dubernet C and Couvreur P. Nanoparticles in cancer therapy and diagnosis (2002) Advanced Drug Delivery Reviews 64: 24-36. https://doi.org/10.1016/S0169-409X(02)00044-3
- Zhang L and Webster TJ. Nanotechnology and nanomaterials: Promises for improved tissue regeneration (2009) Nano Today 4: 66-80. https://doi.org/10.1016/j.nantod.2008.10.014
- Mei L, Zhang Z, Zhao L, Huang L, Yang XL, et al. Pharmaceutical nanotechnology for oral delivery of anticancer drugs (2013) Advanced Drug Delivery Reviews 65: 880-890. https://doi.org/10.1016/j.addr.2012.11.005
- Van Vlerken LE, Vyas TK and Amiji MM. Poly (ethylene glycol)-modified nanocarriers for tumor-targeted and intracellular delivery (2007) Pharmaceutical Research 24: 1405-1414. https://doi.org/10.1007/s11095-007-9284-6
- Musacchio T and Torchilin VP. Recent developments in lipid-based pharmaceutical nanocarriers (2011) Frontiers in Bioscience 16: 1388-1412. https://doi.org/10.2741/3795
- Zhang J, Wang L, You X, Xian T, Wu J, et al. Nanoparticle Therapy for Prostate Cancer: Overview and Perspectives (2019) Current Topics in Medicinal Chemistry 19: 57-73. https://doi.org/10.2174/1568026619666190125145836
- Ganju A, Yallapu MM, Khan S, Behrman SW, Chauhan SC, et al. Nanoways to overcome docetaxel resistance in prostate cancer (2014) Drug Resistance Updates 17: 13-23. https://doi.org/10.1016/j.drup.2014.04.001
- Singh S, Sharma A and Robertson GP. Realizing the clinical potential of cancer nanotechnology by minimizing toxicologic and targeted delivery concerns (2012) Cancer Research 72: 5663-5668. https://doi.org/10.1158/0008-5472.CAN-12-1527
- Arias JL. Drug targeting strategies in cancer treatment: an overview (2010) Mini-Reviews in Medicinal Chemistry 11:1-17. https://doi.org/10.2174/138955711793564024
- Maeda H, Nakamura H and Fang J. The EPR effect for macromolecular drug delivery to solid tumors: Improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in-vivo (2013) Advanced Drug Delivery Reviews 65: 71-79.https://doi.org/10.1016/j.addr.2012.10.002
- Torchilin V. Tumor delivery of macromolecular drugs based on the EPR effect (2011) Advanced Drug Delivery Reviews 63: 131-135. https://doi.org/10.1016/j.addr.2010.03.011
- Barenholz Y. Doxil® - The first FDA-approved nano-drug: Lessons learned (2012) Journal of Controlled Release 160: 117-134. https://doi.org/10.1016/j.jconrel.2012.03.020
- Byrne JD, Betancourt T and Brannon-Peppas L. Active targeting schemes for nanoparticle systems in cancer therapeutics (2008) Advanced Drug Delivery Reviews 60: 1615-1626. https://doi.org/10.1016/j.addr.2008.08.005
- Fenske DB and Cullis PR. Liposomal nanomedicines (2008) Expert Opinion on Drug Delivery 5: 25-44. https://doi.org/10.1517/17425247.5.1.25
- Maurer N, Fenske DB and Cullis PR. Developments in liposomal drug delivery systems (2001) Expert Opinion on Biological Therapy 1: 923-947.https://doi.org/10.1517/14712598.1.6.923
- Zedan AH, Hansen TF, Assenholt J, Pleckaitis M, Madsen JS, et al. MicroRNA expression in tumour tissue and plasma in patients with newly diagnosed metastatic prostate cancer (2018) Tumor Biology 40.https://doi.org/10.1177/1010428318775864
- Kita K and Dittrich C. Drug delivery vehicles with improved encapsulation efficiency: taking advantage of specific drug-carrier interactions (2011) Expert Opinion on Drug Delivery 8: 329-342. https://doi.org/10.1517/17425247.2011.553216
- Qiu Y and Park K. Environment-sensitive hydrogels for drug delivery (2012) Advanced Drug Delivery Reviews 64: 49-60. https://doi.org/10.1016/j.addr.2012.09.024
- Oh JK, Drumright R, Siegwart DJ and Matyjaszewski K. The development of microgels/nanogels for drug delivery applications (2008) Progress in Polymer Science (Oxford) 33: 448-477. https://doi.org/10.1016/j.progpolymsci.2008.01.002
- Ning P, Lü S, Bai X, Wu X, Gao C, et al. High encapsulation and localized delivery of curcumin from an injectable hydrogel (2018) Materials Science and Engineering C 83: 121-129.https://doi.org/10.1016/j.msec.2017.11.022
- Zhao J, Li Y and Wang M. Fabrication of robust transparent hydrogel with stretchable, self-healing, easily recyclable and adhesive properties and its application (2019) Materials Research Bulletin 112: 292-296. https://doi.org/10.1016/j.materresbull.2018.12.033
- Pooresmaeil M and Namazi H. Preparation and characterization of polyvinyl alcohol/β-cyclodextrin/GO-Ag nanocomposite with improved antibacterial and strength properties (2019) Polymers for Advanced Technologies 30: 447-456. https://doi.org/10.1002/pat.4484
- Kankala RK, Kuthati Y, Sie HW, ShihHY, Lue SI, et al. Multi-laminated metal hydroxide nanocontainers for oral-specific delivery for bioavailability improvement and treatment of inflammatory paw edema in mice (2015) Journal of Colloid and Interface Science 458: 217-228. https://doi.org/10.1016/j.jcis.2015.07.044
- Lee SH, Song JG and Han HK. Development of pH-responsive organic-inorganic hybrid nanocomposites as an effective oral delivery system of protein drugs (2019) Journal of Controlled Release 311: 74-84.https://doi.org/10.1016/j.jconrel.2019.08.036
- Vahed AT, Naimi-Jamal MR and Panahi L. Alginate-coated ZIF-8 metal-organic framework as a green and bioactive platform for controlled drug release (2019) Journal of Drug Delivery Science and Technology 49: 570-576. https://doi.org/10.1016/j.jddst.2018.12.022
- Savjani KT, Gajjar AK and Savjani JK. Drug solubility: importance and enhancement techniques (2012) ISRN Pharmaceutics. https://doi.org/10.5402/2012/195727
- Famuyiwa OT, Jebelli J, Diaka JKK and Asghar W. Interaction between 3-bromopyruvate and sc-514 in prostate cancer treatment (2018) Journal of Cancer Prevention and Current Research 9: 270-280.https://doi.org/10.15406/jcpcr.2018.09.00367
- Anwer MK, Mohammad M, Ezzeldin E, Fatima F, Alalaiwe A, et al. Preparation of sustained release apremilast-loaded PLGAlga nanoparticles: In-vitro characterization and in-vivo pharmacokinetic study in rats (2019) International Journal of Nanomedicine 2019: 1587-1595.https://doi.org/10.2147/IJN.S195048
- Govender T, Stolnik S, Garnett MC, Illum L and Davis SS. PLGA nanoparticles prepared by nanoprecipitation: Drug loading and release studies of a water soluble drug (1999) Journal of Controlled Release 57: 171-185.https://doi.org/10.1016/S0168-3659(98)00116-3
- Drummond DC, Noble CO, Guo Z, Hong K, Park JW, et al. Development of a highly active nanoliposomal irinotecan using a novel intraliposomal stabilization strategy (2006) Cancer Research 66: 3271-3277. https://doi.org/10.1158/0008-5472.CAN-05-4007
- Johnston MJW, Semple SC, Klimuk SK, Edwards K, Eisenhardt ML, et al. Therapeutically optimized rates of drug release can be achieved by varying the drug-to-lipid ratio in liposomal vincristine formulations (2006) Biochimica et Biophysica Acta (BBA) Biomembranes 1758: 55-64. https://doi.org/10.1016/j.bbamem.2006.01.009 Joguparthi V and Anderson BD. Liposomal delivery of hydrophobic weak acids: Enhancement of drug retention using a high intraliposomal pH (2008) Journal of Pharmaceutical Sciences 97: 433-454. https://doi.org/10.1002/jps.21135
- Washington C. Drug release from microdisperse systems: a critical review (1990) International Journal of Pharmaceutics 58: 1-12. https://doi.org/10.1016/0378-5173(90)90280-H
- Herman EH, Vicl JA, Rahmar A, Schein PS and Ferrara JV. Prevention of chronic doxorubicin cardiotoxicity in beagles by liposomal encapsulation (1983) Cancer Research 43: 5427-2432.
- Barenholz Y. Relevancy of drug loading to liposomal formulation therapeutic efficacy (2003) Journal of Liposome Research 13: 1-8. https://doi.org/10.1081/LPR-120017482
- Washington C and Koosha F. Drug release from microparticulates; deconvolution of measurement errors (1990) International Journal of Pharmaceutics 59: 79-82 https://doi.org/10.1016/0378-5173(90)90067-E
- Lorenzo CA and Concheiro A. Smart drug delivery systems: from fundamentals to the clinic (2014) Chemical Communications 50: 7743:7765. https://doi.org/10.1039/c4cc01429d
- Bao B, Thakur A, Li Y, Ahmad A, Azmi AS, et al. The immunological contribution of nf-κb within the tumor microenvironment: a potential protective role of zinc as an anti-tumor agent (2012) Biochimica et Biophysica Acta - Reviews on Cancer 1825: 160-172.https://doi.org/10.1016/j.bbcan.2011.11.002
- Park M and Hong J. Roles of nf-κb in cancer and inflammatory diseases and their therapeutic approaches (2016) Cells 5: 15. https://doi.org/10.3390/cells5020015
- Zhou J, Ching YQ and Chng WJ. Aberrant nuclear factor-kappa B activity in acute myeloid Leukemia: From molecular pathogenesis to therapeutic target (2015) Oncotarget 6: 5490-5500. https://doi.org/10.18632/oncotarget.3545
- deGraffenried LA, Chandrasekar B, Friedrichs WE, Donzis E, Silva J, et al. NF-κB inhibition markedly enhances sensitivity of resistant breast cancer tumor cells to tamoxifen (2004) Annals of Oncology 15: 885-890.https://doi.org/10.1093/annonc/mdh232
- Crowell JA, Steele VE, Sigman CC and Fay JR. Is inducible nitric oxide synthase a target for chemoprevention? (2003) Molecular Cancer Therapeutics 2: 815-823.
- Pautz A, Art J, Hahn S, Nowag S, Voss C, et al. Regulation of the expression of inducible nitric oxide synthase (2010) Nitric Oxide - Biology and Chemistry 23: 75-93.https://doi.org/10.1016/j.niox.2010.04.007
- Dolcet X, Llobet D, Pallares J and Matias-Guiu X. NF-kB in development and progression of human cancer (2005) Virchows Archiv 446: 475-482.https://doi.org/10.1007/s00428-005-1264-9
- SA, Zaitseva L, Langa S, Bowles KM and MacEwan DJ. Flip regulation of ho-1 and tnf signalling in human acute myeloid leukemia provides a unique secondary anti-apoptotic mechanism (2010) Oncotarget 1: 359-366.https://doi.org/10.18632/oncotarget.168
- Gyrd-Hansen M and Meier P. IAPs: From caspase inhibitors to modulators of NF-κB, inflammation and cancer (2010) Nature Reviews Cancer 10: 561-574. https://doi.org/10.1038/nrc2889
- Li X, Abdel-Mageed AB, Mondal D and Kandil E. The nuclear factor kappa-B signaling pathway as a therapeutic target against thyroid cancers (2013) Thyroid 23: 209-218. https://doi.org/10.1089/thy.2012.0237
- Carbone C and Melisi D. NF-κB as a target for pancreatic cancer therapy (2012) Expert Opinion on Therapeutic Targets 16: 1-10. https://doi.org/10.1517/14728222.2011.645806
- Jain G, Cronauer MV, Schrader M, Möller P and Marienfeld RB. NF-κB signaling in prostate cancer: A promising therapeutic target? (2012) World Journal of Urology 30: 303-310. https://doi.org/10.1007/s00345-011-0792-y
- Hjortso M and Andersen M. The expression, function and targeting of haem oxygenase-1 in cancer (2014) Current Cancer Drug Targets 14: 337-347.https://doi.org/10.2174/1568009614666140320111306
- Jeney V, Balla J, Yachie A, Varga Z, Vercellotti GM, et al. Pro-oxidant and cytotoxic effects of circulating heme (2002) Blood 100: 879-887. https://doi.org/10.1182/blood.V100.3.879
- Caballero B. The global epidemic of obesity: An overview (2007) Epidemiologic Reviews 29: 1-5.https://doi.org/10.1093/epirev/mxm012
- Ogden CL, Carroll MD, Kit BK and Flegal KM. Prevalence of obesity in the United States, 2009-2010 (2012b) NCHS Data Brief 82: 1-8.
- Hales CM, Carroll MD, Fryar CD and Ogden CL. Prevalence of obesity among adults and youth: united states, 2015-2016 (2017) NCHS Data Brief 288: 1-8
- Flegal KM, Kruszon-Moran D, Carroll MD, Fryar CD and Ogden CL. Trends in obesity among adults in the United States, 2005 to 2014 (2016) JAMA - Journal of the American Medical Association 315: 2284-2291. https://doi.org/10.1001/jama.2016.6458
- Chooi YC, Ding C and Magkos F. The epidemiology of obesity (2019) Metabolism: Clinical and Experimental 92: 6-10. https://doi.org/10.1016/j.metabol.2018.09.005
- Helmchen LA and Henderson RM. Changes in the distribution of body mass index of white US men, 1890-2000 (2004) Annals of Human Biology 31: 174-181. https://doi.org/10.1080/03014460410001663434
- Flegal KM, Carroll MD, Ogden CL and Johnson CL. Prevalence and trends in obesity among US adults, 1999-2000 (2002) JAMA 288: 1723-1727. https://doi.org/10.1001/jama.288.14.1723
- Flegal KM, Carroll MD, Ogden CL and Curtin LR. Prevalence and trends in obesity among US adults, 1999-2008 (2010) JAMA. https://doi.org/10.1001/jama.2009.2014
- Ogden CL, Carroll MD, Curtin LR, McDowell MA, Tabak CJ, et al. Prevalence of overweight and obesity in the United States, 1999-2004 (2006) JAMA 295: 1549-1555. https://doi.org/10.1001/jama.295.13.1549
- Flegal KM, Carroll D, Kit BK and Ogden CL. Prevalence of obesity and trends in the distribution of body mass index among US adults, 1999-2010 (2012) JAMA 307: 491-497. https://doi.org/10.1001/jama.2012.39
- Hedley AA, Ogden CL, Johnson CL, Carroll MD, Curtin LR, et al. Prevalence of overweight and obesity among US children, adolescents, and adults, 1999-2002 (2004) Journal of the American Medical Association 291: 2847-2850. https://doi.org/10.1001/jama.291.23.2847
- Ogden CL, Carroll MD, Kit BK and Flegal KM. Prevalence of obesity and trends in body mass index among US children and adolescents, 1999-2010 (2012a) JAMA 307: 483-490. https://doi.org/10.1001/jama.2012.40
- McAllister EJ, Dhurandhar NV, Keith SW, Aronne LJ, Barger J, et al. Ten putative contributors to the obesity epidemic (2009) Critical Reviews in Food Science and Nutrition 49: 868-913. https://doi.org/10.1080/10408390903372599
- Ogden CL, Fakhouri TH, Carroll MD, Hales CM, Fryar CD, et al. Prevalence of Obesity Among Adults, by Household Income and Education - United States, 2011-2014 (2017) MMWR Morbidity and Mortality Weekly Report 66: 1369-1373. https://doi.org/10.15585/mmwr.mm6650a1
- Amling CL, Kane CJ, Riffenburgh RH, Ward JF, Roberts JL, et al. Relationship between obesity and race in predicting adverse pathologic variables in patients undergoing radical prostatectomy (2001) Urology 58: 723-728. https://doi.org/10.1016/s0090-4295(01)01373-5
- Mallah KN, DiBlasio CJ, Rhee AC, Scardino PT and Kattan MW. Body mass index is weakly associated with, and not a helpful predictor of, disease progression in men with clinically localized prostate carcinoma treated with radical prostatectomy (2005) Cancer 103: 2030-2034.https://doi.org/10.1002/cncr.20991
- Yamamoto H, Kuno Y, Sugimoto S, Takeuchi H and Kawashima Y. Surface-modified PLGA nanosphere with chitosan improved pulmonary delivery of calcitonin by mucoadhesion and opening of the intercellular tight junctions (2005) J of Controlled Release 102: 373-381.https://doi.org/10.1016/j.jconrel.2004.10.010
- Prego C, García M, Torres D and Alonso MJ. Transmucosal macromolecular drug delivery (2005) J of Controlled Release 101: 151-162. https://doi.org/10.1016/j.jconrel.2004.07.030
- Stella B, Arpicco S, Peracchia MT, Desmaële D, Hoebeke J, et al. Design of folic acid-conjugated nanoparticles for drug targeting (2000) J of Pharma Sci. 89: 1452-1464 https://doi.org/10.1002/1520-6017(200011)89:11<1452::AID-JPS8>3.0.CO;2-P
- Zhang N, Chittasupho C, Duangrat C, Siahaan TJ and Berkland C. PLGA nanoparticle-peptide conjugate effectively targets intercellular cell-adhesion molecule-1 (2008) Bioconjugate Chem 19: 145-152.https://doi.org/10.1021/bc700227z
- Magenheim B, Levy MY and Benita S. A new in-vitro technique for the evaluation of drug release profile from colloidal carriers - ultrafiltration technique at low pressure (1993) International J of Pharmaceutics 94: 115-123.https://doi.org/10.1016/0378-5173(93)90015-8
- Douer D. Efficacy and safety of vincristine sulfate liposome injection in the treatment of adult acute lymphocytic leukemia (2016) The Oncologist 21: 840-847. https://doi.org/10.1634/theoncologist.2015-0391
- Wang D, Kong L, Wang J, He X, Li X, et al. Polymyxin E sulfate-loaded liposome for intravenous use: Preparation, lyophilization, and toxicity assessment in-vivo (2009) PDA Journal of Pharmaceutical Science and Technology 63: 159-167.
- Cui J, Li C, Deng Y, Wang Y and Wang W. Freeze-drying of liposomes using tertiary butyl alcohol/water cosolvent systems (2006) International Journal of Pharmaceutics 312: 131-136. https://doi.org/10.1016/j.ijpharm.2006.01.004
- Boyd BJ. Characterisation of drug release from cubosomes using the pressure ultrafiltration method (2003) International J of Pharmaceutics 260: 239-247.https://doi.org/10.1016/S0378-5173(03)00262-X
- Zhang YV, Wei B, Zhu Y, Zhang Y and Bluth MH. Liquid Chromatography-Tandem Mass Spectrometry: An Emerging Technology in the Toxicology Laboratory (2016) Clinics in Laboratory Medicine 36: 635-661.https://doi.org/10.1016/j.cll.2016.07.001
- Jones G and Kaufmann M. Vitamin D metabolite profiling using liquid chromatography-tandem mass spectrometry (LC–MS/MS) (2016) J of Steroid Biochem and Mol Biology 164: 110-114. https://doi.org/10.1016/j.jsbmb.2015.09.026
- Yadav P, Rath G, Sharma G, Singh R and Goyal AK. Polysorbate 80 coated solid lipid nanoparticles for the delivery of temozolomide into the brain (2018) The Open Pharmacology Journal 8: 21-28.https://doi.org/10.2174/1874143601808010021
- Li B, Wang X, Liu W and Xue Q. Tribochemistry and antiwear mechanism of organic-inorganic nanoparticles as lubricant additives (2006) Tribology Letters 22: 79-84.https://doi.org/10.1007/s11249-005-9002-7
- Jifen W, Wensheng Z and Guifen J. Preparation and tribological properties of tungsten disulfide hollow spheres assisted by methyltrioctylammonium chloride (2010) Tribology International 43: 1650-1658.https://doi.org/10.1016/j.triboint.2010.03.012
- Yang G Bin, Chai ST, Xiong XJ, Zhang SM, Yu LG, et al. Preparation and tribological properties of surface modified Cu nanoparticles (2012) Transactions of Nonferrous Metals Society of China 22: 366-372. https://doi.org/10.1016/S1003-6326(11)61185-0
- Chen Y, Zhang Y, Zhang S, Yu L, Zhang P, et al. Preparation of nickel-based nanolubricants via a facile in situ one-step route and investigation of their tribological properties (2013) Tribology Letters 51: 73-83. https://doi.org/10.1007/s11249-013-0148-4
- Xiao-kun M, Lee NH, Oh HJ, Kim JW, Rhee CK, et al. Surface modification and characterization of highly dispersed silica nanoparticles by a cationic surfactant (2010) Colloids and Surfaces A: Physicochemical and Engineering Aspects 358: 172-176. https://doi.org/10.1016/j.colsurfa.2010.01.051
- Ishii F and Nagasaka Y. Simple and Convenient Method for Estimation of Marker Entrapped in Liposomes (2001) Journal of Dispersion Science and Technology 22: 970-101. https://doi.org/10.1081/DIS-100102684
- Marshall J. Transwell ® invasion assays (2011) Methods in Molecular Biology 769: 97-110.https://doi.org/10.1007/978-1-61779-207-6_8
- Bian Y, Du Y, Wang R, Chen N, Du X, et al. A comparative study of HAMSCs/HBMSCs transwell and mixed coculture systems (2019) IUBMB Life 71: 1048-1055. https://doi.org/10.1002/iub.2074
- Li C, Zheng J, Chen S, Huang B, Li G, et al. RRM2 promotes the progression of human glioblastoma (2018) Journal of Cellular Physiology 233: 6759-6767.https://doi.org/10.1002/jcp.26529
- Schachner M, Wortham KA, Ryberg MZ, Dorfman S and Campbell GLM. Brain cell surface antigens detected by anticorpus callosum antiserum (1977) Brain Research 127: 87-97. https://doi.org/10.1016/0162-0134(86)80048-4
- Famuyiwa TO. A new approach for preparing sc-514 loaded plga particles by single emulsion method (2019) Journal of Medical Pharmaceutical and Allied Sciences 8: 2367-2380. https://doi.org/10.22270/jmpas.v8i6.872
- Alfarouk KO, Stock CM, Taylor S, Walsh M, Muddathir AK, et al. Resistance to cancer chemotherapy: Failure in drug response from ADME to P-gp (2015) Cancer Cell International 15. https://doi.org/10.1186/s12935-015-0221-1
- Ruman U, Fakurazi S, Masarudin MJ and Hussein MZ. Nanocarrier-based therapeutics and theranostics drug delivery systems for next generation of liver cancer nanodrug modalities (2020) International Journal of Nanomedicine 2020: 1437-1456. https://doi.org/10.2147/IJN.S236927
- Masood F. Polymeric nanoparticles for targeted drug delivery system for cancer therapy (2016) Materials Science and Engineering C 60: 569-578.https://doi.org/10.1016/j.msec.2015.11.067
- Kang B, Zheng MB, Song P, Chen AP, Wei JW, et al. Subcellular-Scale Drug Transport via Ultrasound-Degradable Mesoporous Nanosilicon to Bypass Cancer Drug Resistance (2017) Small 13. https://doi.org/10.1002/smll.201604228
- Mondal L, Mukherjee B, Das K, Bhattacharya S, Dutta D, et al. CD-340 functionalized doxorubicin-loaded nanoparticle induces apoptosis and reduces tumor volume along with drug-related cardiotoxicity in mice (2019) International Journal of Nanomedicine 14: 8073-8094. https://doi.org/10.2147/IJN.S220740
- Abd Ellah NH and Abouelmagd SA. Surface functionalization of polymeric nanoparticles for tumor drug delivery: approaches and challenges (2017) Expert Opinion on Drug Delivery 14: 201-214.https://doi.org/10.1080/17425247.2016.1213238
- Urimi D, Agrawal AK, Kushwah V and Jain S. Polyglutamic Acid Functionalization of Chitosan Nanoparticles Enhances the Therapeutic Efficacy of Insulin Following Oral Administration (2019) AAPS PharmSciTech 20.https://doi.org/10.1208/s12249-019-1330-2
- Swami R, Singh I, Jeengar MK, Naidu VGM, Khan W, et al. Adenosine conjugated lipidic nanoparticles for enhanced tumor targeting (2015) International Journal of Pharmaceutics 486: 287-296. https://doi.org/10.1016/j.ijpharm.2015.03.065
- Song CX, Labhasetwar V, Murphy H, Qu X, Humphrey WR, et al. Formulation and characterization of biodegradable nanoparticles for intravascular local drug delivery (1997) Journal of Controlled Release 43: 197-212. https://doi.org/10.1016/S0168-3659(96)01484-8
- Azevedo CRD, Stosch VM, Costa MS, Ramos AM, Cardoso MM, et al. Modeling of the burst release from PLGA micro and nanoparticles as function of physicochemical parameters and formulation characteristics (2017) International Journal of Pharmaceutics 532: 229-240. https://doi.org/10.1016/j.ijpharm.2017.08.118
- Maulvi FA, Patil RJ, Desai AR, Shukla MR, Vaidya RJ, et al. Effect of gold nanoparticles on timolol uptake and its release kinetics from contact lenses: In-vitro and in-vivo evaluation (2019) Acta Biomaterialia 86: 350-362. https://doi.org/10.1016/j.actbio.2019.01.004
- Borghi A, Foa E, Balossino R, Migliavacca F and Dubini G. Modelling drug elution from stents: Effects of reversible binding in the vascular wall and degradable polymeric matrix (2008) Comp Meth Biomech Biomed Eng 11: 367-377. https://doi.org/10.1080/10255840801887555
- Huang X and Brazel CS. On the importance and mechanisms of burst release in matrix-controlled drug delivery systems (2001) J Controlled Release 73: 121-136. https://doi.org/10.1016/S0168-3659(01)00248-6
- Loew S, Fahr A and May S. Modeling the Release Kinetics of poorly water-soluble drug molecules from liposomal nanocarriers (2011) J Drug Delivery. https://doi.org/10.1155/2011/376548
- Zeng L, An L and Wu X. Modeling drug-carrier interaction in the drug release from nanocarriers (2011) J Drug Delivery. https://doi.org/10.1155/2011/370308
- Jelvehgari M, Valizadeh H, Rezapour M and Nokhodchi A. Control of encapsulation efficiency in polymeric microparticle system of tolmetin (2010) Pharm Develop Techn 15: 71-79. https://doi.org/10.3109/10837450903002173
- Yeo Y and Park K. Control of encapsulation efficiency and initial burst in polymeric microparticle systems (2004) Archives of Pharmacal Research.
- Shukla S, MacLennan GT, Fu P, Patel J, Marengo SR, et al. Nuclear factor-κB/p65 (Rel A) is constitutively activated in human prostate adenocarcinoma and correlates with disease progression (2004) Neoplasia 6: 390-400. https://doi.org/10.1593/neo.04112
- Ghantous A, Sinjab A, Herceg Z and Darwiche N. Parthenolide: From plant shoots to cancer roots (2013) Drug Discovery today 18: 894-905.https://doi.org/10.1016/j.drudis.2013.05.005
- Pacifico F and Leonardi A. NF-κB in solid tumors (2006) Biochemical Pharmacology 72: 1142-1152.https://doi.org/10.1016/j.bcp.2006.07.032
- Karin M and Greten FR. NF-κB: Linking inflammation and immunity to cancer development and progression (2005) Nature Reviews Immunology. https://doi.org/10.1038/nri1703
- Hua X, Tan S, Bandara HMHN, Fu Y, Liu S, et al. Externally controlled triggered-release of drug from PLGA micro and nanoparticles (2014) PLoS ONE.
- Bhagav P, Upadhyay H and Chandran S. Brimonidine tartrate-eudragit long-acting nanoparticles: Formulation, optimization, in-vitro and in-vivo evaluation (2011) AAPS PharmSciTech. https://doi.org/10.1208/s12249-011-9675-1
- Ünal S, Aktaş Y, Benito JM and Bilensoy E. Cyclodextrin nanoparticle bound oral camptothecin for colorectal cancer: Formulation development and optimization (2020) Inte J Pharma 584: 119468.https://doi.org/10.1016/j.ijpharm.2020.119468
- Kaleemuddin M and Srinivas P. Lyophilized oral sustained release polymeric nanoparticles of nateglinide (2013) AAPS PharmSciTech. https://doi.org/10.1208/s12249-012-9887-z
- Sethi M, Sukumar R, Karve S, Werner ME, Wang EC, et al. Effect of drug release kinetics on nanoparticle therapeutic efficacy and toxicity (2014) Nanoscale https://doi.org/10.1039/c3nr05961h
- Song B. Lotus leaf-inspired design of calcium alginate particles with superhigh drug encapsulation efficiency and pH responsive release (2018) Colloids and Surfaces B: Biointerfaces 172: 464-470.https://doi.org/10.1016/j.colsurfb.2018.09.001
- Qaddoumi MG, Gukasyan HJ, Davda J, Labhasetwar V, Kim KJ, et al. Clathrin and caveolin-1 expression in primary pigmented rabbit conjunctival epithelial cells: Role in PLGA nanoparticle endocytosis (2003) Molecular Vision.
- Palocci C, Valletta A, Chronopoulou L, Donati L, Bramosanti M, et al. Endocytic pathways involved in PLGA nanoparticle uptake by grapevine cells and role of cell wall and membrane in size selection (2017) Plant Cell Reports. https://doi.org/10.1007/s00299-017-2206-0
- Meyer MC and Guttman DE. Dynamic dialysis as a method for studying protein binding I: Factors affecting the kinetics of dialysis through a cellophane membrane (1970a) J Pharma Sci 59: 33-38. https://doi.org/10.1002/jps.2600590104
- Meyer MC and Guttman DE. Dynamic dialysis as a method for studying protein binding II: Evaluation of the method with a number of binding systems (1970b) J Pharma Sci 59: 39-48. https://doi.org/10.1002/jps.2600590105
- Zambito Y, Pedreschi E and Di Colo G. Is dialysis a reliable method for studying drug release from nanoparticulate systems? - A case study (2012) Int J Pharma 434: 28-34. https://doi.org/10.1016/j.ijpharm.2012.05.020
- Khaira R, Sharma J and Saini V. Development and characterization of nanoparticles for the delivery of gemcitabine hydrochloride (2014) The Scientific World Journal. https://doi.org/10.1155/2014/560962
- Modi S and Anderson BD. Determination of drug release kinetics from nanoparticles: Overcoming pitfalls of the dynamic dialysis method (2013) Molecular Pharmaceutics. https://doi.org/10.1021/mp400154a
- Wallace SJ, Li J, Nation RL and Boyd BJ. Drug release from nanomedicines: Selection of appropriate encapsulation and release methodology (2012) Drug Delivery and Translational Research. https://doi.org/10.1007/s13346-012-0064-4
- Semete B, Booysen L, Lemmer Y, Kalombo L, Katata L, et al. In-vivo evaluation of the biodistribution and safety of PLGA nanoparticles as drug delivery systems (2010) Nanomedicine: Nanotechnology, Biology, and Medicine 6: 662-671. https://doi.org/10.1016/j.nano.2010.02.002
- Navarro SM, Morgan TW, Astete CE, Stout RW, Coulon D, et al. Biodistribution and toxicity of orally administered poly (lactic-co-glycolic) acid nanoparticles to F344 rats for 21 days (2016) Nanomedicine 11: 1653-1669. https://doi.org/10.2217/nnm-2016-0022
- Niessen WMA. Liquid chromatography | Mass spectrometry (2019) In Encyclopedia of Analytical Science https://doi.org/10.1016/B978-0-12-409547-2.142131
- Perreault H and Lattová E. Mass spectrometry (2019) In Comprehensive Biotechnology 1: 679-687. https://doi.org/10.1016/B978-0-444-64046-8.00039-2
- Berthiller F, Werner U, Sulyok M, Krska R, Hauser MT, et al. Liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) determination of phase II metabolites of the mycotoxin zearalenone in the model plant Arabidopsis thaliana (2006) Food Additives and Contaminants 23: 1194-1200. https://doi.org/10.1080/02652030600778728
- van de Merbel AF, van der Horst G, Buijs JT and van der Pluijm G. Protocols for migration and invasion studies in prostate cancer (2018) In Methods in Molecular Biology 1786: 67-79. https://doi.org/10.1007/978-1-4939-7845-8_4
- Karbarz M, Mytych J, Solek P, Stawarczyk K, Tabecka-Lonczynska A, et al. Cereal grass juice in wound healing: hormesis and cell-survival in normal fibroblasts, in contrast to toxic events in cancer cells (2019) Journal of Physiology and Pharmacology : An Official Journal of the Polish Physiological Society 70: 595-604. https://doi.org/10.26402/jpp.2019.4.10
- Gupta A, Behl T, Heer HR, Deshmukh R and Sharma PL. Mdm2-P53 interaction inhibitor with cisplatin enhances apoptosis in colon and prostate cancer cells in-vitro (2019) Asian Pacific Journal of Cancer Prevention 20: 3341-3351. https://doi.org/10.31557/APJCP.2019.20.11.3341
- Collak FK, Demir U and Sagir F. YAP1 Is Involved in Tumorigenic Properties of Prostate Cancer Cells (2020) Pathology and Oncology Research 26: 867-876. https://doi.org/10.1007/s12253-019-00634-z
- Darwish NHE, Sudha T, Godugu K, Bharali DJ, Elbaz O, et al. Novel targeted nano-parthenolide molecule against NF-kB in acute myeloid leukemia (2019) Molecules 24: 2103. https://doi.org/10.3390/molecules24112103
- Zarabi MF, Farhangi A, Mazdeh SK, Ansarian Z, Zare D, et al. Synthesis of gold nanoparticles coated with aspartic acid and their conjugation with FVIII protein and FVIII antibody (2014) Indian Journal of Clinical Biochemistry 29: 154-160. https://doi.org/10.1007/s12291-013-0323-2
- Adamo G, Campora S and Ghersi G. Functionalization of nanoparticles in specific targeting and mechanism release (2017) In Nanostructures for Novel Therapy: Synthesis, Characterization and Applications 57-80.https://doi.org/10.1016/B978-0-323-46142-9.00003-7
- Thiruppathi R, Mishra S, Ganapathy, Padmanabhan P and Gulyás B. Nanoparticle functionalization and its potentials for molecular imaging (2017) Advanced Science 4 https://doi.org/10.1002/advs.201600279
- Preecha P and Jettanasen J. Investigation of functionalized silicon nanoparticles by size exclusion chromatography (2017) Materials Research Express 4. https://doi.org/10.1088/2053-1591/aa6638
Corresponding author
Toluleke
Oloruntobi Famuyiwa,
Department
of Biological Sciences, Florida Atlantic University, 777 Glades Road, Boca
Raton, FL 33431, USA,
E-mail: famuyiwatoluleke@gmail.com
Citation
Famuyiwa
TO, Bowers Z, Bentley A, Caraballo D, Subtil P, et al. Drug release studies of SC-514
PLGA nanoparticles (2021) Pharmacovigil and Pharmacoepi 4: 1-21.